

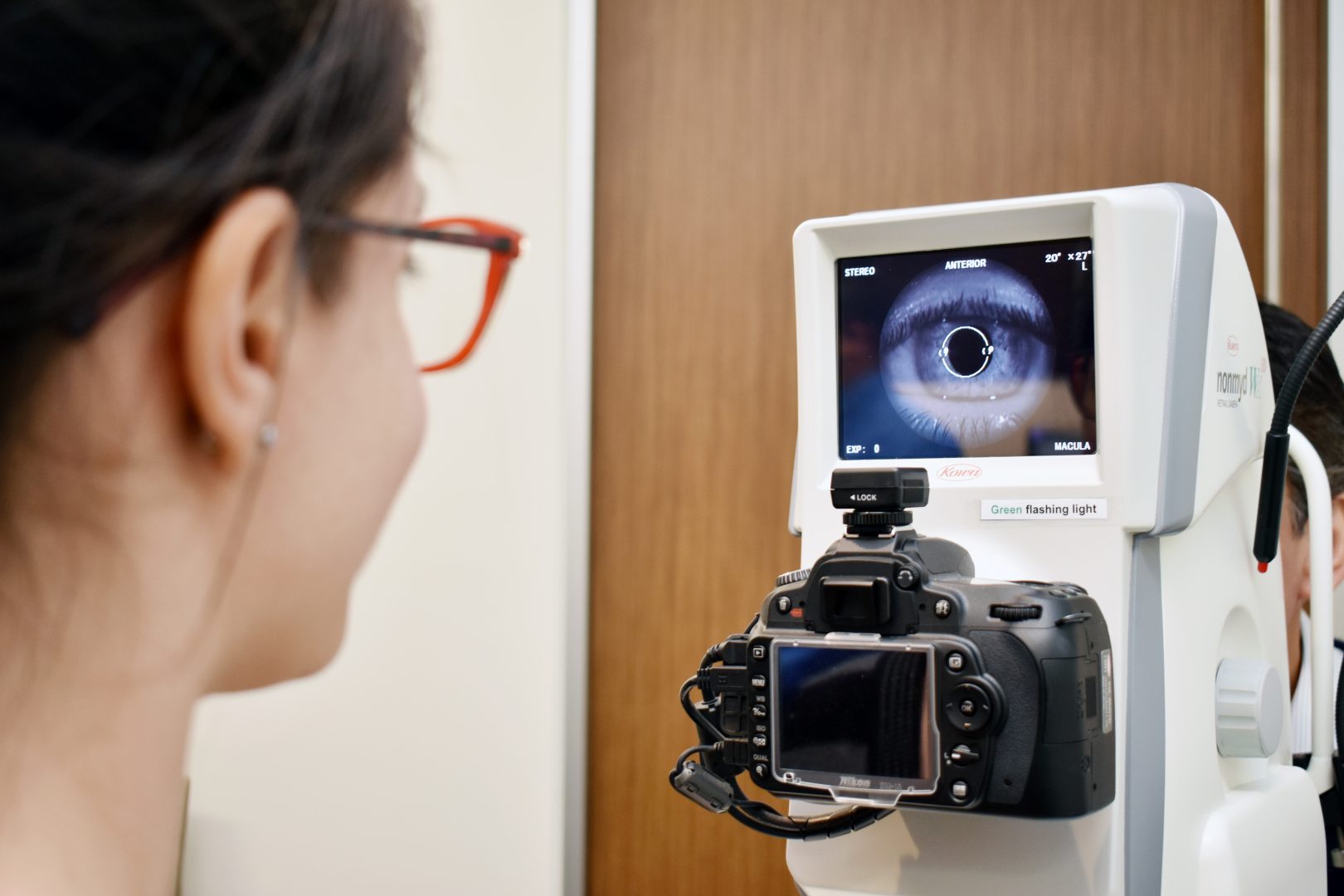


- Refer a Patient
- Referral Types
- Patient Information
- Overview of CFEH Clinics
- CFEH Instrument List
- Our clinical team
- Causes of Vision Loss
- Patient Forms












Facebook Cases
Facebook Case 156
Case ##156 PDF Download
For the original question click here
Answer
In June 2019, CFEH introduced a new Management option for referring optometrists where CFEH takes on the management of the patient’s referred condition only and patients are returned to their referring practitioner for all other ocular needs. In this case, the referring optometrist had selected the Management option thus the CFEH clinical team were able to discuss the diagnosis with the patient, organise a timely follow up with a glaucoma specialist ophthalmologist, discuss treatment options, facilitate the patient’s access to treatment and also conduct the follow up care of this patient. This allowed the patient timely access to appropriate care and saved the referring practitioner valuable time in organising on-referral and follow-up but maintain primary care for all other aspects.
Facebook case #155
Case ##155 PDF Download
For the original question click here
Answer
This patient has iridociliary cysts which are causing the iris to be pushed anteriorly, narrowing the angle.
)
CFEH Facebook Case #153
Case #153 PDF Download
For the original question click here
Answer
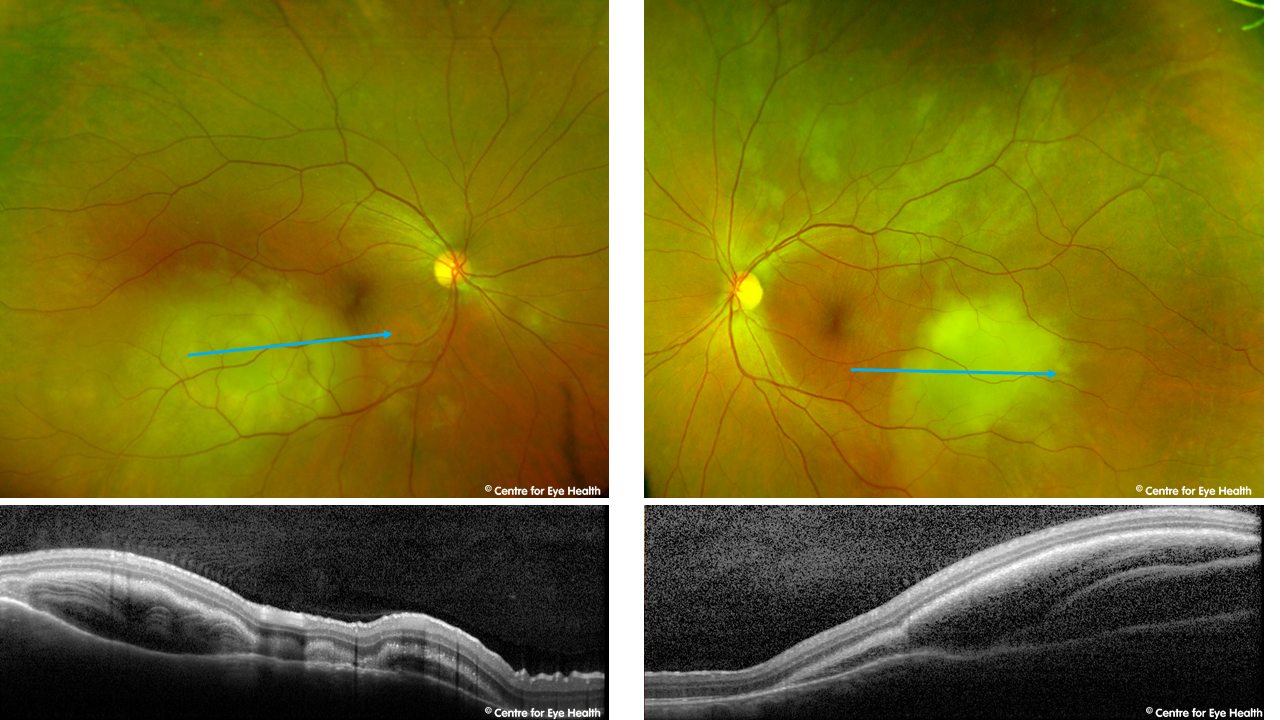
The Spectralis near infrared reflectance image outlines a sharply demarcated, hypo-reflective, wedge –shaped lesion with its apex pointing towards the fovea. The lesion corresponds to the shape and location of the scotoma mapped out on Amsler. OCT B -scan shows focal outer retinal disturbance with increased hyper-reflectivity in the outer nuclear layer and external limiting membrane as well as disturbance of the ellipsoid zone.
This presentation is consistent with a diagnosis of acute macular neuroretinopathy in the left eye.
Acute macular neuroretinopathy is a rare condition of unknown cause that is most commonly found in young white females. It is commonly associated with non-specific flu-like illness, fever, or the use of oral contraceptives.
This condition is characterised by small paracentral lesions with associated scotomas and may affect one or both eyes. The lesions are initially best seen on infra-red but become more visible over days to weeks on examination and retinal photography. OCT imaging through the lesions usually show disruption of the ellipsoid zone.
CFEH Facebook case #152
Case #152 PDF Download
For the original question click here
Answer
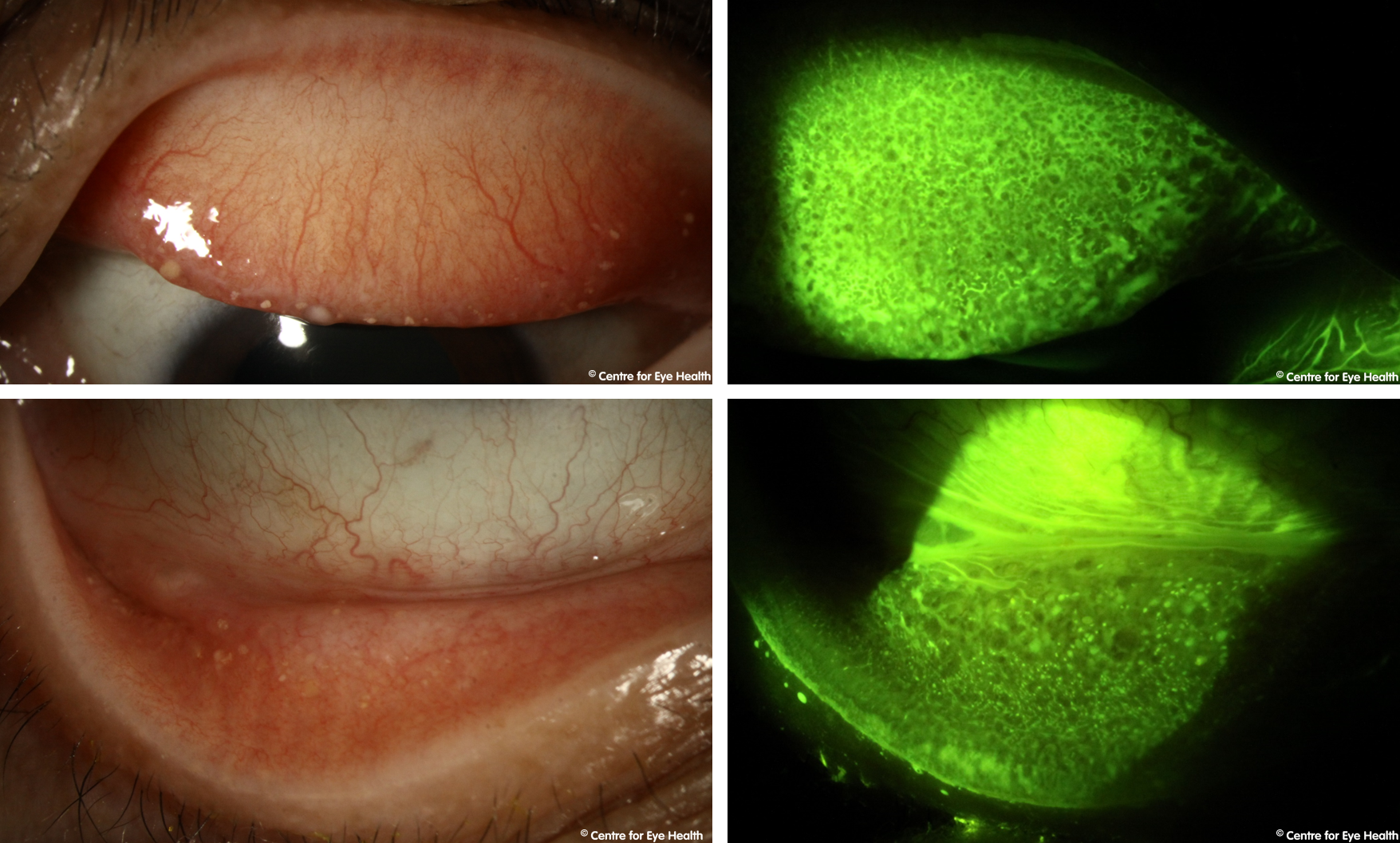
Trachoma is a bacterial infection caused by Chlamydia trachomatis. It is typically associated with poor hygiene and/or poverty and is easily spread through direct or indirect contact with infected eye secretions. If left untreated, trachoma can cause blindness with chronic, recurrent inflammation causing scarring of the palpebral conjunctiva which leads to entropian, trichiasis and corneal scarring.
Although improved hygiene has seen the eradication of this disease in many countries of the world, it is still found in more than 50 developing countries throughout Africa and Asia. Australia is the only developed country where this condition may still be seen – typically in remote outback communities. Progress is being made towards the eradication of this disease in Australia, a goal expected to be met in the near future.
Infection starts with conjunctivitis and lid oedema due to inflammation. Early signs of infection include the presence of lid follicles and papillae which undergo progressive hypertrophy. Corneal pannus starts to develop (the growth of blood vessels past the limbus and into the cornea). Over time symblephron form, lids become entropic and the resulting trichiasis causes corneal scarring and vision impairment.
)
CFEH Facebook Case #151
Case #151 PDF Download
For the original question click here
Answer
The right disc is irregular in shape and enlarged with a grey oval excavation noted at the inferior margin. There is a complete absence of neuro-retinal rim inferonasally and OCT imaging through the disc shows a deep cavity inferiorly. A peripapillary retinoschisis can also be seen inferiorly.
These findings are consistent with a diagnosis of optic disc coloboma. This is a congenital condition which may be inherited (autosomal dominant or autosomal recessive) or may be an isolated anomaly.
Colobomas form due to the incomplete closure of the embryonic fissure during foetal development. They are typically found inferonasally and there may be an associated field defect. Colobomas may occur in the retina/choroid, optic nerve, iris, or in combination, so a thorough ocular examination should be conducted to rule out involvement in other locations. In this case the coloboma was limited to the optic disc.
CFEH Facebook Case #150
Case #150 PDF Download
For the original question click here
Answer
A 52 year old Caucasian female presented for examination of peripheral retinal lesions. She has a history of breast cancer that was treated 12 months previously with surgery and multiple rounds of chemotherapy. Pupils were equal, round and reactive and the anterior segment normal. Posterior widefield images and OCT scans are below. The lesions are seen to be elevated with significant sub-retinal fluid present. The presentation is consistent with a diagnosis of choroidal metastasis. These findings should be communicated promptly to the patient’s oncologist.
)
Case #149
Case #149 PDF Download
For the original question click here
Answer
This patient has a retinal detachment in the superior nasal retina. An epiretinal membrane is an unusual finding in a 28 year old which prompted our optometrist to look more closely at this case.
The detachment is very difficult to see using just the 2 dimensional widefield image alone,but is more obvious when viewed in 3-D during a routine dilated retinal examination. This highlights the importance of a thorough peripheral retinal examination, supplemented by multi-modal imaging where possible.
The fundus autofluorescence image gives us a hint that there is something unusual in this case with a hyper-AF line present at the borer of the detachment. Confirmation is provided by OCT imaging through this area.
)
CFEH Facebook Case #148
Case #148 PDF Download
For the original question click here
Answer
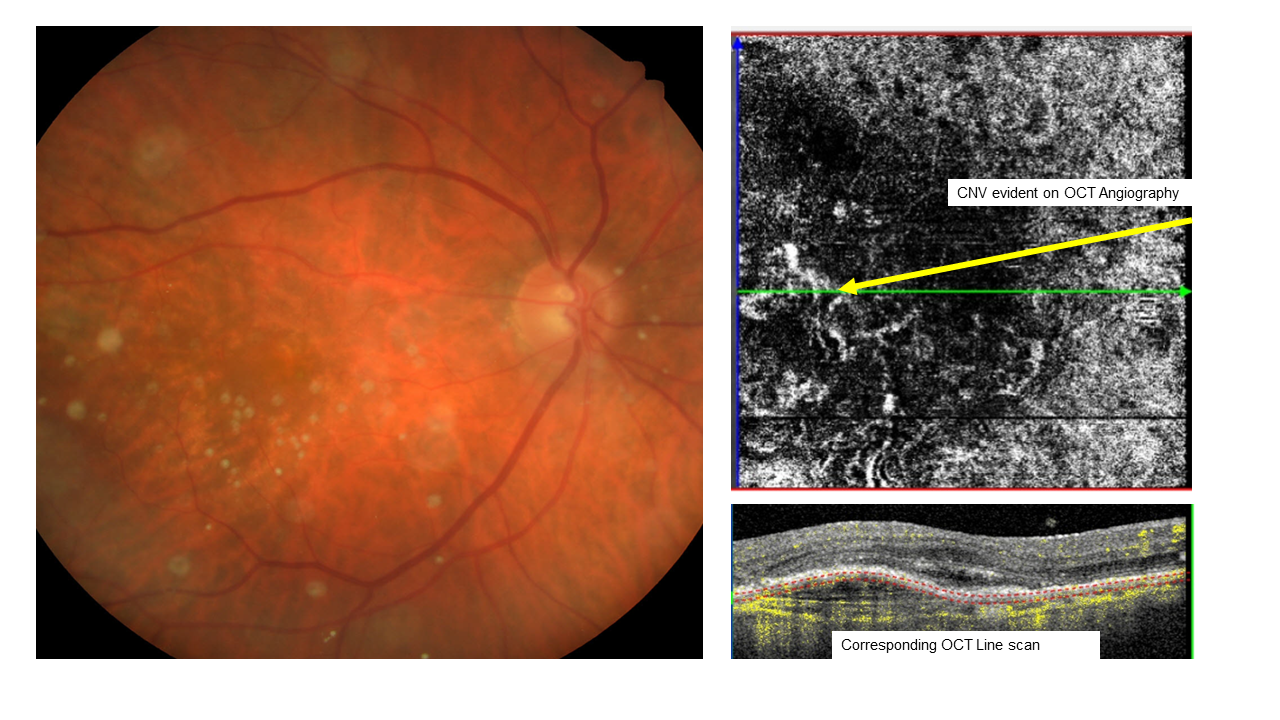
Focal choroidal excavation (FCE) refers to a localised region of excavation within the choroid. While the aetiology of this finding is not completely understood, it has been associated with pathologies where the choriocapillaris is dysfunctional, including central serous chorioretinopathy (CSCR) and polypoidal choroidal vasculopathy (PCV).
This has recently led some authors to consider FCE as part of the pachychoroid spectrum of diseases (Chung et al. 2017 Retina). Similar to many conditions in the pachychoroid spectrum of disease, a potential sequela of FCE is choroidal neovascularisation (CNV). While the pathophysiology of this
process is not yet completely understood, FCE should be considered a risk factor for CNV and patients should be followed up regularly to monitor for this development.
)
CFEH Facebook Case #147
Case #147 PDF Download
For the original question click here
Answer
These are OCTA images of the macula showing the superficial blood vessels of the macula.
Image 2 is a 60-year-old with type 2 diabetes and poor glycaemic control.
A few key features of OCT angiography include:
Prominent microaneurysms (circled)
Foveal avascular zone enlargement and (red square)
A loose (reduced density) capillary network demonstrating reduced capillary perfusion (a few examples in green arrows)
This presentation is consistent with early macular ischaemia.
)
CFEH Facebook Case #146
Case #146 PDF Download
For the original question click here
Answer
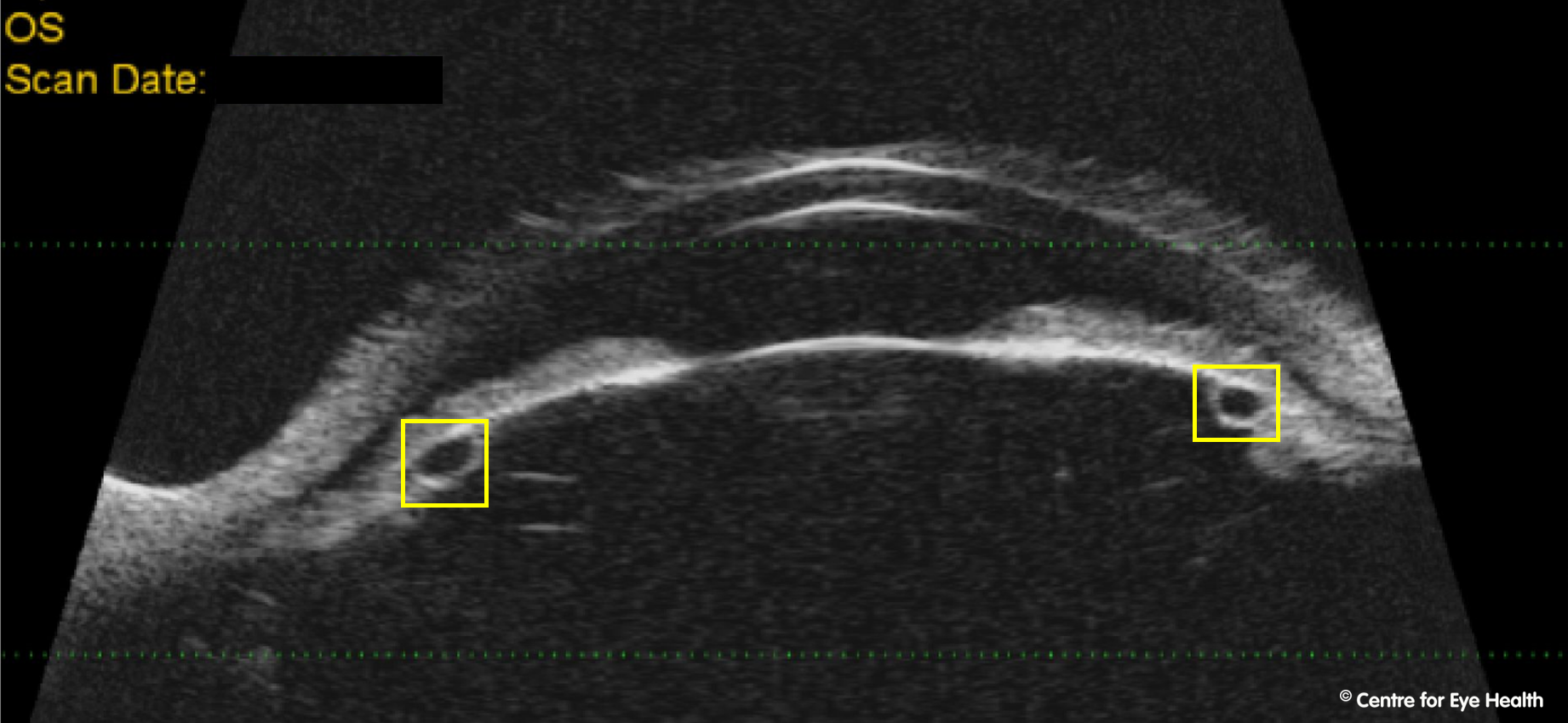
Optomap imaging shows irregular yellow lesions located superotemporally near the vascular arcades in each eye. OCT imaging through the lesions show the lesions to have an irregular contour and the masses are shown to originating from the sclera, causing thinning of the overlying choroid and also associated RPE abnormalities. These findings are consistent with a diagnosis of sclerochoroidal calcification – a diagnosis confirmed with B-scan ultrasound which showed a hyper-echoic area with posterior shadowing in each eye.
Sclerochoroidal calcifications can be either unilateral or bilateral and are described as yellow placoid lesions with defined borders, typically found in the superotemporal equatorial retina. They are most commonly found in elderly Caucasian males. Most cases are idiopathic, however this condition can also be associated with abnormal calcium-phosphorus metabolism or renal tubular hypokalemic metabolic alkalosis syndromes.
Due to the possibility of systemic associations with these lesions, this patient was referred to an ophthalmologist for assessment.
Facebook case #144
Case #144 PDF Download
For the original question click here
Answer
The Spectralis HRA2+OCT has several different imaging modalities that can be used to highlight different pathologies.
- Multicolour imaging: generates a series of three images simultaneously using different laser wavelengths (blue, green, infrared), providing additional information on the appearance of distinct structures at different depths within the retina. This imaging modality can be used on patients with media opacities or nystagmus and may highlight pathologies not readily visible with a traditional retinal examination. A 2017 study by Graham et al, showed multicolour to be more sensitive than colour retinal photography for detecting signs of early AMD.
- Infrared reflectance: uses a longer wavelength of light to illuminate the fundus. This has several advantages as it can more readily penetrate opaque media (eg due to cataract) and the RPE, thus revealing structures deep in the choroid. Clinical uses include assessing the macula for reticular pseudodrusen in age-related macular degeneration.
- Blue reflectance: “red-free” imaging which is achieved through illumination of the retina with blue light. These images are particularly useful to highlight pathologies affecting the superficial retina including retinal folds, epiretinal membranes and the retinal nerve fibre layer defects.
- Green reflectance: is achieved through retinal illumination with green light and is most useful for examining blood, blood vessels, and exudates
Facebook case #143
Case ##143 PDF Download
For the original question click here
Answer
The OCT line scan in the first image shows an irregular serous pigment epithelial detachment (PED)containing some hyer-reflective material inferior to the fovea. The fundus autofluorescence image shows two areas of hyper-autofluorescence with adjacent mottled areas of hypo-autoflourescence and relative hypo-autofluorescence surrounding the fovea. The en face OCT image through the choroid shows enlarged choroidal vessels while the OCTA image of the avascular complex shows a network of irregular vessels, presumed to be choroidal neovascularisation. These clinical signs are consistent with a diagnosis of pachychoroid neovasculopathy (PNV).
One common feature seen in PNV is the presence of the “double layer sign”- a shallow, irregular elevation of the RPE, separating it from the underlying intact Bruch’s membrane. This appears as two highly reflective layers: one at the level of the RPE and another beneath the RPE and can be seen in an additional OCT line scan in our patient (below).
PNV is part of the pachychoroid spectrum of disease; characterised by dilated/enlarged choroidal vessels (often corresponding to a thickened choroid) and associated with progressive RPE dysfunction. This disease spectrum includes pachychoroid epitheliopathy, central serous chorioretinopathy, PNV and polypoidal choroidal vasculopathy/aneurysmal type 1 neovascularization.
The underlying mechanism causing pachychoroidal changes is postulated to be microtrauma of Bruch’s membrane due to the enlargement of vessels in Haller’s layer of the choroid. This results in attenuation of the choriocapillaris and subsequent RPE changes which can progress to neovascularization in the later stages.
This patient was referred to an ophthalmologist for further investigation.
)
Facebook case #142
Case #142 PDF Download
For the original question click here
Answer
Spectralis OCT revealed inner retinal cavitation and outer retinal thinning at the fovea in both eyes. OCT Angiography demonstrates temporal juxtafoveal microaneurysms and telangiectatic vessels, more notable in the right eye. These changes are consistent with a diagnosis of macular telangiectasia type 2.
The pathogenesis of Mac Tel 2 is poorly understood, current thought is that it is a primary neuro retinal degenerative condition with a second vascular involvement. It is relatively rare with a prevalence of 0.1% in the general population based on grading of stereoscopic fundus photographs. This is likely to be underestimated as OCT and fundus autofluorescence imaging that can detect disease at an earlier stage was not performed.
Symptoms typically start in the 5th or 6th decade of life and the early signs and symptoms may be very subtle but are usually limited to mildly reduced visual acuity and metamorphopsia which increase as the disease progresses. Typical clinical findings include a dulling or loss of the foveal reflex, a greying of the parafoveolar area, small foveal cystoid changes and pigment clumping. A pseudo-vitelliform lesion can also form in the central macula.
This patient is in the non-proliferative stage of this disease as there are no clinical signs of sub-retinal neovascularisation and fibrosis, consequently he was asked to return for a 6 month on-going review. While there is no treatment for the pre-proliferative stages, photodynamic therapy may be useful in decreasing vision loss in the proliferative stages.
Facebook case #141
Case ##141 PDF Download
For the original question click here
Answer
Retinal photos show a very pale, cupped nerve in the right eye and minimal RNFL reflectivity – findings that indicate right optic atrophy. It is likely that this is the result of the trauma sustained 6 years ago. While the metal object did not penetrate the globe, the damage to the lower lid and the patient’s description of the accident indicate that the metal spoke could have travelled between the globe and the orbital wall causing a forceful rotation of the globe.
This type of force can result in optic nerve avulsion whereby there is a traumatic disinsertion of the nerve fibres at the disc margin (the disc sheath remains undamaged) leading to a rare form of traumatic optic neuropathy. In the acute phase, other clinical signs associated with this condition may include peripapillary subretinal or vitreous haemorrhage and choroidal folds due to peripapillary retinal oedema.
While it is not expected that the structural damage or visual field defect will be progressive, a review was scheduled for 6 months to ascertain this.
Facebook case #140
Case #Case #140 PDF Download
For the original question click here
Answer
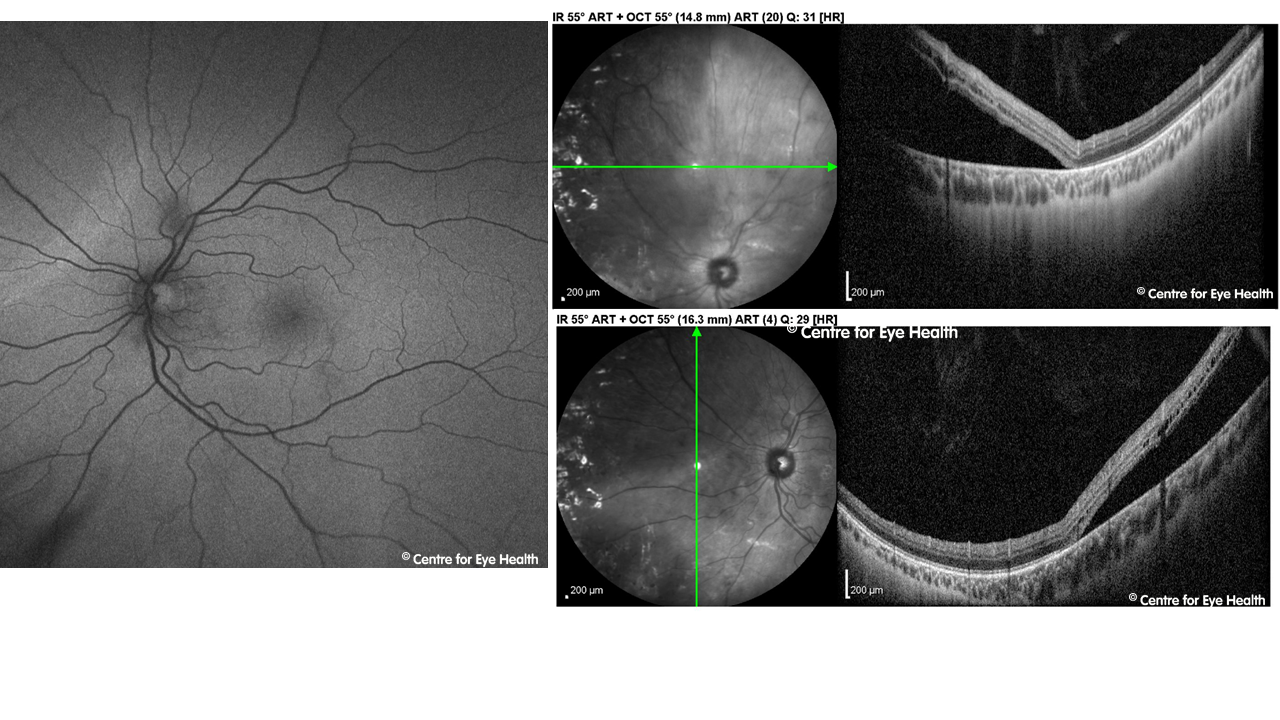
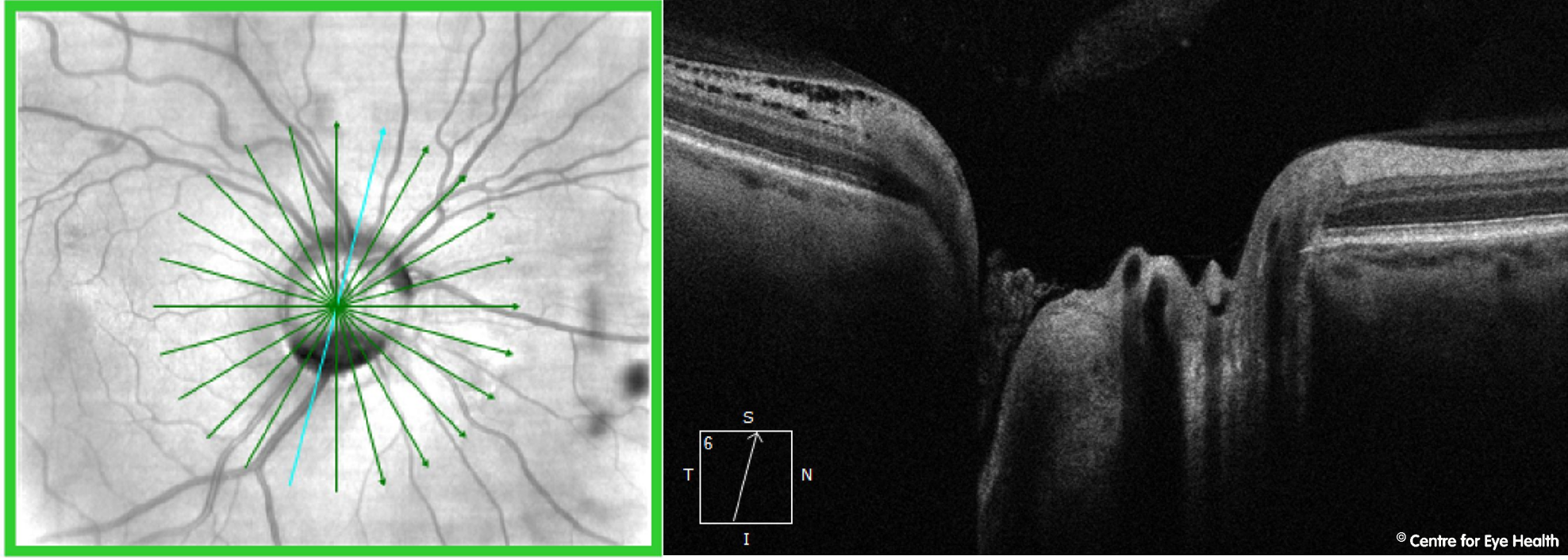
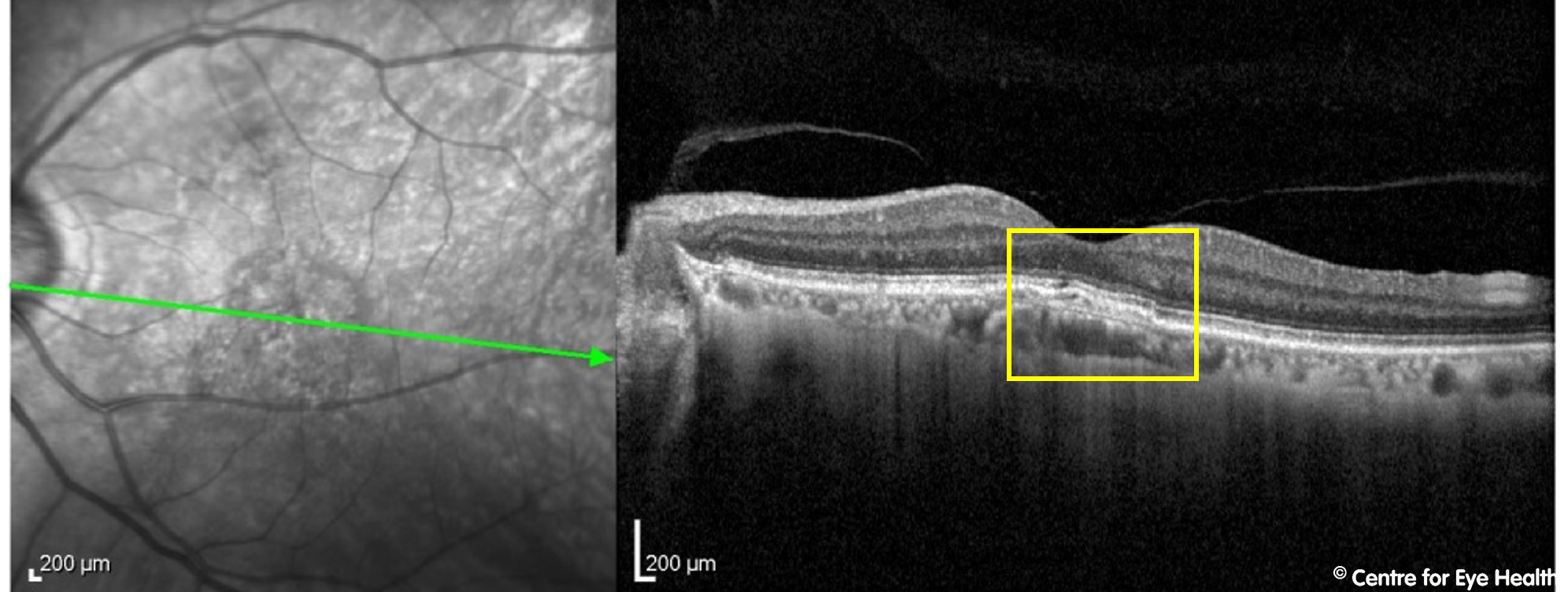
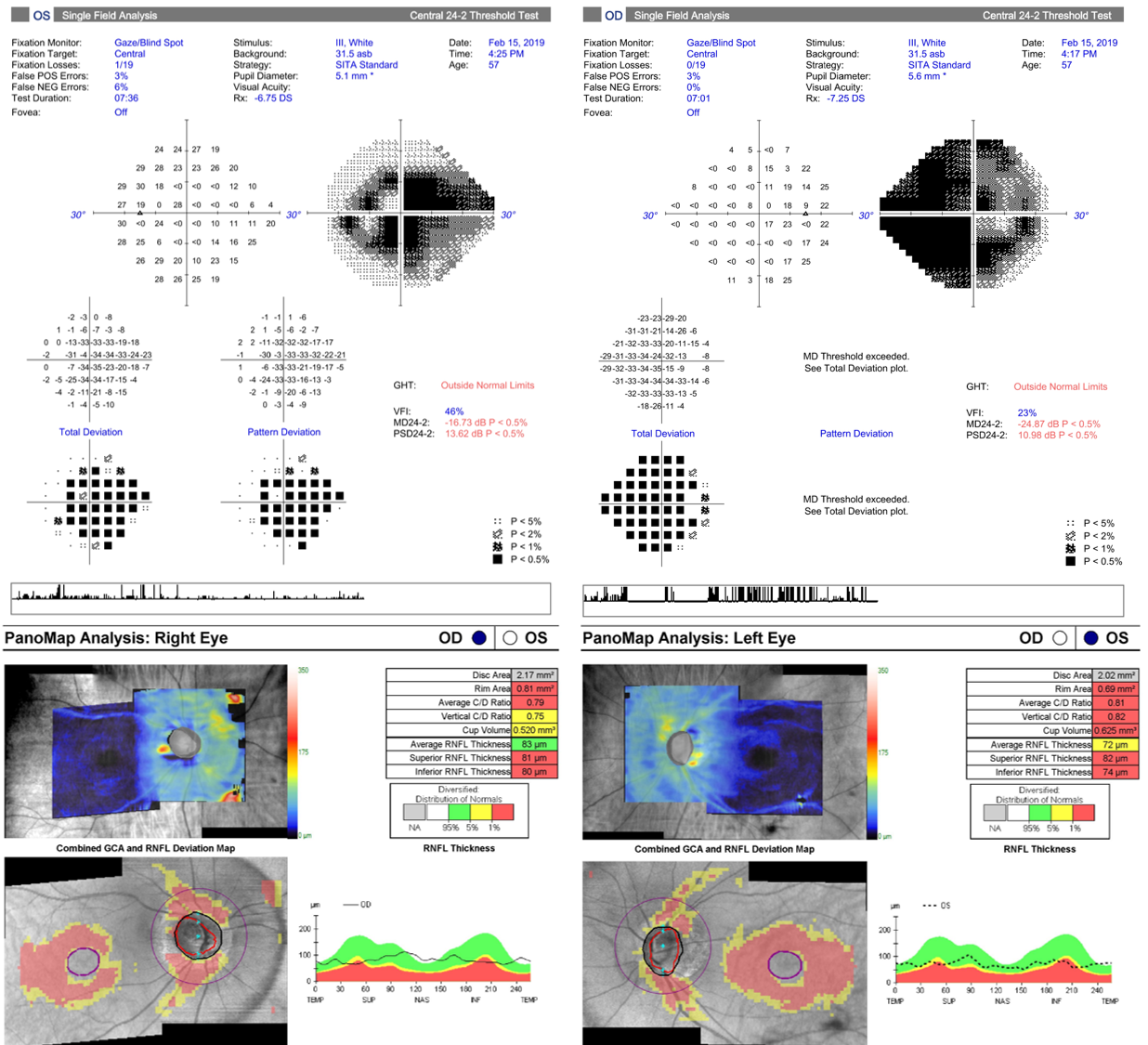
While this patient has a retinoschisis superonasal to right disc and a peripapillary intrachoroidal cavitation inferior to the left disc as seen on Spectralis line scans, these are not the cause of potential vision loss. This patient has advanced glaucoma.
This case is an example of how high myopia can make glaucoma difficult to detect. The NRR is difficult to assess given the asymmetric and myopic nature of the discs, however it appeared notably thin both superiorly and inferiorly in both eyes. The OCT results shown below should be viewed with caution due to questionable segmentation from the high myopia, however results suggest a very thin RNFL in both eyes. GCA values should be viewed with caution due to the high myopia affecting the normative comparisons as well as the right staphyloma affecting the segmentation.
The visual fields results reveal typically glaucomatous field defects that are concordant with the structural changes indicated on OCT analysis. The mean deviation in both eyes indicate that this is a case of advanced bilateral glaucoma. This patient is outside the scope of glaucoma that is treatable at CFEH by co-management so this patient was referred to a glaucoma specialist.
)
Facebook case #139
Case ##139 PDF Download
For the original question click here
Answer
Endothelial cell profiles of the right eye show substantial polymegethism and associated reduction in endothelial cell density over a period of 7 years. Endothelial cell density does decrease naturally with age, however several studies show that both posterior chamber phakic IOLs and iris-fixated IOLs, as per this case, can also contribute to reduced endothelial cell density. While the studies vary significantly in terms of the extent of this reduction in cell density, it has been reported to be as high as 10.5% 22 months post-surgery compared to pre-surgical measurements (Koss and Kohnen, 2009).
Facebook case #138
Case ##138 PDF Download
For the original question click here
Answer
The OCT line scan taken through the anomalous vessel shows a hyper-reflective area in the anterior choroid with posterior shadowing. The RPE is slightly elevated in this area and the extended depth capabilities of the OCT instrument show that the vessel has a large diameter in this area. The OCTA choroidal raster scan confirms the location of the vessel to be choroidal. There is an area of hyper-autofluoresence at the location of the anomalous vessel.
There are several differentials for this clinical finding:
- It may be a congenital macrovessel. These are larger and more tortuous than normal retinal vessels and perfuse a larger area of retina. These vessels are more likely to develop vascular occlusive disease
- The vessel may be an anomalous posterior ciliary vessel
- Various nematodes (worms) have been documented to produce sub-retinal atrophic or cicatricial tracts in the fundus similar to what is seen on the Optomap image from our patient.
Based on the OCT findings, it is most likely that this is a case of choroidal macrovessel, however confirmation with fluorescein angiography is advisable.
Facebook case #137
Case #137 PDF Download
For the original question click here
Answer
On examination, the right eye was found to have a dark foveal reflex and OCT shows a small area of disruption of the photoreceptors and RPE at the fovea centralis. These findings indicate the presence of a macular microhole.
Macular microholes are often an incidental finding in an asymptomatic patient. Less commonly these patients may present with mildly reduced VA, scotoma and/or metamorphosia.
Abnormal vitreomacular interaction is a well-known inciting factor for macular microholes. Similar clinical findings can also be caused by sun-gazing (solar retinopathy), watching an eclipse without protective eyewear, the use of recreational drugs (alkyl nitrite compounds, also known as “poppers”) and blunt trauma however this patient denied any history of these.
Microholes are generally benign and vision would typically be expected to be well-preserved. With this in mind and taking into account the unknown etiology of the microhole, the patient was recommended for review in 12 months.
Facebook Case #136
Case #136 PDF Download
For the original question click here
Answer
The disc photos show bitemporal pallor but no significant cupping or thinning of the neuro-retinal rim that would be typical of glaucoma. The visual field defects are not typical of glaucoma, with no structure-function concordance with the OCT results.
GCA showed bi-nasal thinning of the ganglion cell layers, with areas of thinning respecting the vertical midline. This pattern of thinning is suspicious of neurological damage, and, in this case, the cause is retrograde degeneration secondary to neurological damage
The key clinical messages here are that Structure-function discordance is a suspicious feature in optic nerve disease, especially in the absence of characteristic glaucomatous changes at the optic disc. In cases where GCA thinning respects the vertical midline, neurological causes should be excluded.
Facebook Case #135
Case #135 PDF Download
For the original question click here
Answer
The iris lesion is darkly pigmented and elevated with iridocorneal contact with no sentinel vessels visible. Anterior OCT showed significant iris elevation, iridocorneal contact and also corneal involvement (stroma and endothelial layers). These findings are consistent with a diagnosis of long-standing primary pigment epithelial cyst.
Primary iris pigment epithelial cysts are cavities that arise between the iris pigment epithelial layers. They can occur at the pupillary margin, in the mid-iris or peripherally in the iridociliary sulcus, as in our case. They may also be found free-floating in the anterior chamber or vitreous. Most iris pigment epithelal cysts are non-progressive and rarely cause secondary ocular complications.
An ultrasound biomicroscopy examination has been scheduled to confirm this diagnosis and provide a baseline for future review. A 6 month review was recommended to establish stability.
Facebook case #134
Case #134 PDF Download
For the original question click here
Answer
The haemorrhage is a Roth spot, characterised by the white spot in the centre.
Roth spots are thought to result from the rupture of retinal capillaries and extrusion of blood, leading to the formation of a platelet-fribin thrombus (the white spot). historically these were first identified in patients with acute bacterial endocartitis and for some time were considered pathognomonic of this condition.
Retinal haemorrhages are commonly seen in patients with underlying systemic disease processes that predispose to retinal endothelial dysfunction and rupture including diabetes, hypertension, anaemia, bacterial endocarditis and HIV but they can also be seen in cases of significant trauma or intracranial haemorrhage.
The patient was referred to his GP for a full systemic workup.
Facebook case #133
Case #133 PDF Download
For the original question click here
Answer
The fundus autofluorescence image shows an area of mottled hyper and hypo-AF just inferior to the superior vascular arcades . An OCT scan through the region shows thinning of the outer retinal layers (circled below) with a hyper reflective lesion likely representing pigment migration and also enlarged choroidal vessels (arrows below).
These changes are consistent with pachychoroid spectrum disease, in this case most likely a prior episode of central serous chorioretinopathy.
The lower OCT image in this patient shows a “saw-tooth” appearance to the drusen – a feature characteristic of cuticular drusen. The image provided shows the difference in appearance between the 2 drusen types. Cuticular drusen are located between the retinal pigment epithelium (RPE) and Bruch’s membrane like hard and soft drusen, however there are several significant differences. Cuticular drusen are typically small in diameter like hard drusen but are more numerous, often coalescing. They typically have steeply sloping sides and have been referred to in the literature as having a “saw-toothed” appearance and are characterised on fluorescein angiography by a “starry-sky” appearance. Several characteristics of cuticular drusen are similar to those of soft drusen – coalescence, resorption and RPE disturbances (Balaratnasingam et al 2017) and the 2 drusen sub-types share comparable risk of progression to late AMD (geographic atrophy or neovascular AMD).
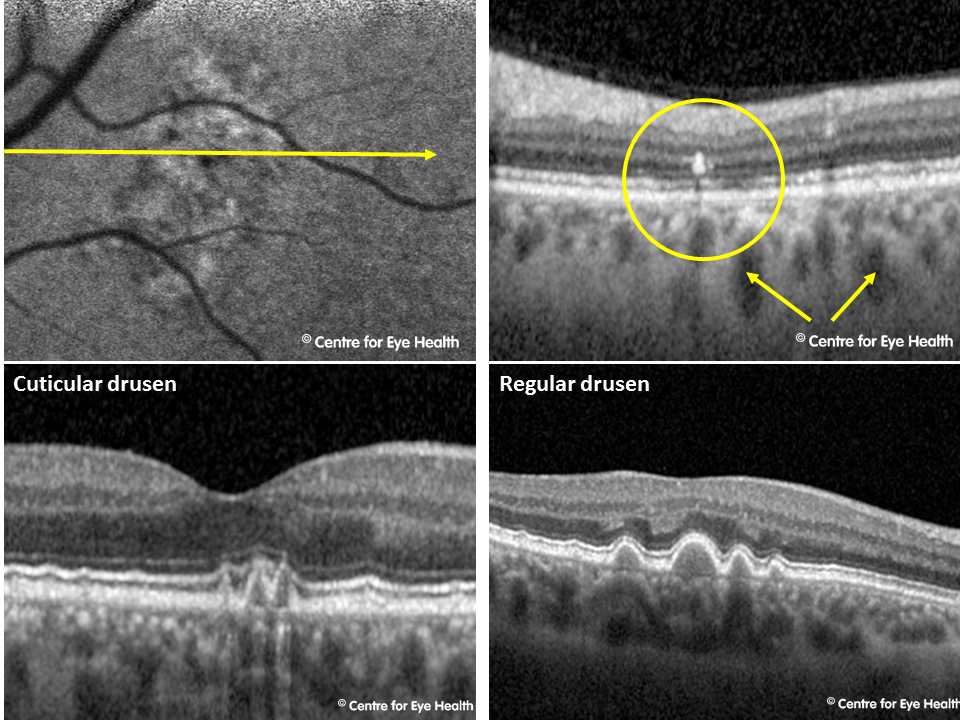)
Facebook Case #132
Case #132 PDF Download
For the original question click here
Answer
This patient has bilateral disc-like areas of stromal haze in the central cornea. OCT scans show focal areas of irregular thickening and thinning as well as increased hyper-reflectivity of the posterior cornea. Corneal topography showed anterior corneal steepening superonasally with flattening of the inferotemporal cornea. Central corneal thicknesses were greater than average at 633µm (OD) and a similar reading on the left eye.
These findings are consistent with a diagnosis of posterior keratoconus – a rare, non-progressive condition with sporadic presentation. It is usually considered a developmental disorder, however it may also arise secondary to ocular trauma. Vision impairment may arise from stromal scarring or amblyopia.
There are 2 distinct types of this condition described in the literature:
- Keratoconus posticus generalis: characterised by increased curvature of the entire posterior cornea
- Keratoconus posticus cirumscriptus: characterised by localized paracentral or central posterior corneal indentation, as in our patient.
This patient was scheduled for review in 6 months to establish stability of the condition.
Facebook case #131
Case #131 PDF Download
For the original question click here
Answer
In this patient’s retinal photo, subtle darkly pigmented lines may be seen around the posterior pole. These extend from the peripapillary region out to the superior and inferior vascular arcades. There are numerous areas of hypo-autofluoresence seen along both vascular arcades, and on OCT areas of focal RPE thickening can be seen. This presentation is consistent with a diagnosis of reticular dystrophy of the RPE which is a type of pattern dystrophy.
Early presentation of this condition is characterised by pigmentary changes which start at the fovea and move outwards from there. This pigment eventually fades, leaving RPE atrophy in its place. Fortunately vision is usually minimally affected, even in advanced cases, however in some cases atrophy and choroidal neovascularisation can cause vision loss.
Associated ocular conditions may include spherophakia with myopia and luxated lens, partial iris atrophy, scleral staphyloma, convergent strabismus, and choroidal neovascularisation. There may also be systemic associations, including deaf-mutism and choreatiform behaviour (involuntary movement disorder).
An annual review is recommended, and family members should also be screened as this condition is hereditary.
Facebook Case #130
Case #130 PDF Download
For the original question click here
Answer
This patient has significant optic atrophy with the Panomap showing marked loss of the RNFL and Ganglion cell complex. The location of loss matches the areas of reduced RNFL reflectivity on red-free and is concordant with the visual field defects noted in this eye. The cause of the optic atrophy may be glaucoma, however given the asymmetric presentation, this patient was sent for neuro-imaging to eliminate other potential causes of optic neuropathy.
The OCT line scan showed small elongated hypo-reflective spaces in the INL consistent with microcystic macular oedema (MME) secondary to optic atrophy. This condition is differentiated from traditional cystoid macular oedema in that no leakage is found with fluorescein angiography.
MMO has only been recently reported in the literature and was initially believed to be a finding specific to multiple sclerosis-related optic neuropathy. Since then, MME has been reported in a range of conditions causing optic atrophy or optic neuritis, including Leber’s hereditary optic neuropathy, advanced glaucoma and optic nerve head drusen. The exact causative mechanism is still the subject of much debate however studies have shown that the prevalence of MME is increased with more severe RNFL/GCL thinning, indicating it is a marker for disease severity.
There is no known treatment for MME and recent studies show no consistency with regards to progression – some will improve, others worsen and some will remain stable, however there have been no documented changes to visual acuity as a result of MME.
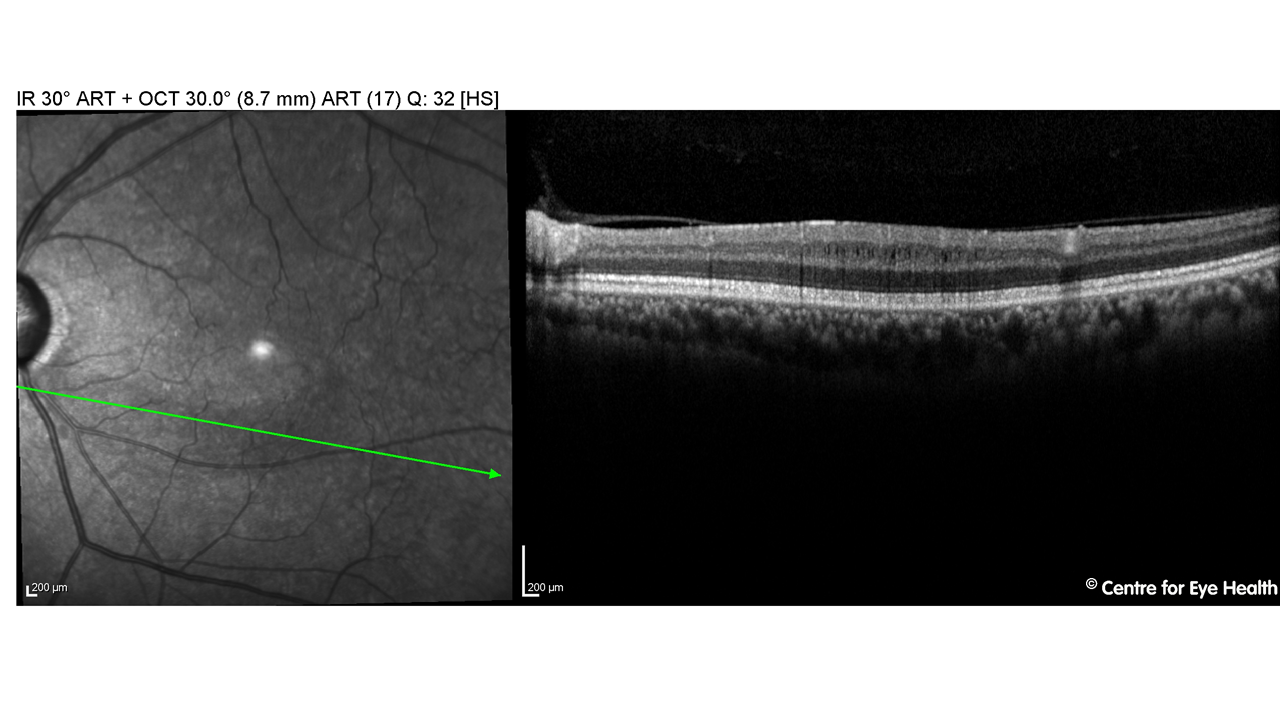)
CFEH Facebook Case #129
Case #129 PDF Download
For the original question click here
Answer
Anterior eye imaging revealed hypertrophy of the iris pigment epithelium across the anterior iris surface, nasally in the right eye and superiorly in the left. Optovue OCT and UBM scans showed increased hyper-reflectivity of these lesions. They appeared to arise from the posterior margin of the iris, with no associated mass of cystic changes posterior to the lesions.
This appearance is consistent with a diagnosis of mild ectropian uveae (EU) which is defined as the presence of iris pigment epithelium on the anterior iris surface.
EU can be either congenital or acquired. Acquired cases may be caused by iris neovascularisation / neovascular glaucoma, or they may be secondary to inflammatory, ischemic, or neoplastic conditions affecting the iris. Acquired EU are usually progressive due to membranous iris traction.
Congenital cases of EU are rare and non-progressive but are often associated with systemic conditions including (most commonly) neurofibromatosis 1, but also Prader-Willi syndrome, Rieger anomaly, and facial hemihypertrophydysgenesis. Congenital EU is associated with anterior insertion of the iris, dysgenesis of the drainage angle, and has a high rate of glaucoma (up to 90% of cases develop glaucoma) so these patients should be monitored carefully.
CFEH Facebook Case #128
Case #128 PDF Download
For the original question click here
Answer
An 83 year old female presented for a glaucoma assessment and it was noted that she had a very festive-looking anterior asteroid hyalosis.
Asteroid hyalosis is a degenerative condition characterized by yellow-white spherical deposits of lipid and calcium which usually float in the vitreous without impacting vision. Rarely, the vitreous can prolapse into the anterior chamber in pseudophakic elderly patients and the asteroid hyalosis will appear in the anterior chamber as has happened with this patient.
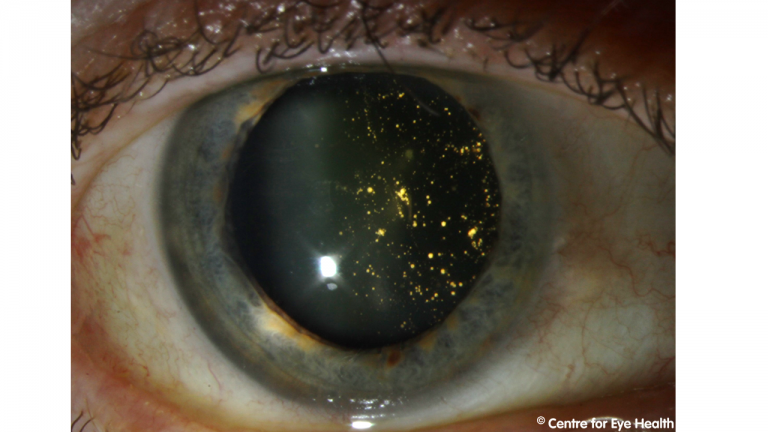)
CFEH Facebook Case #127
Case #127 PDF Download
For the original question click here
Answer
Imaging shows neovascularisation, unusual vessel looping and arteriolar sheathing in the superonasal peripheral retina of the left eye. An OCT scan through this area reveals neovascularisation at the posterior vitreous interface.
These changes indicate a likely systemic cause, however this is not immediately identifiable from an ocular examination alone. The patient was referred to an ophthalmologist and a full systemic workup recommended.
CFEH Facebook Case #126
Case #126 PDF Download
For the original question click here
Answer
The white lesions in the peripheral retina are focal clumps of exudative material. Looking at the magnified red-free image given, there are also microaneurysms and teleangiectatic vessels present in the peripheral retina. These retinal changes most likely reflect peripheral retinal non-perfusion and are likely related to the patient’s high myopia. The differential diagnosis in this patient would be a mild form of familial exudative vitreoretinopathy.
CFEH Facebook Case #125
Case #125 PDF Download
For the original question click here
Answer
A 57 year old female with type 2 diabetes presented for a 12 month routine review. She claims to have good control of her diabetes and takes Diabex and Diamicron, however is unsure of her HbA1c or her blood glucose levels. She mentions that lately she has noticed numbness in her hands. Below are her images today and her images from 12 months ago. Her diabetic retinopathy has progressed significantly and there is non-macula retinal oedema present. She was referred back to her endocrinologist for review and possible discussion of Fenofibrate which may reduce retinopathy progression in patients with mild or moderate NPDR and Type 2 diabetes.
)
CFEH Facebook Case #124
Case #124 PDF Download
For the original question click here
Answer
The lesions seen in this patient are keratic precipitates and are a manifestation of the patient’s sarcoidosis. The incidence of ocular sarcoidosis is unclear with different studies reporting it to be between 13% and 79% in patients with systemic sarcoidosis. Ocular sarcoidosis is characterised by granulomatous inflammation of one or more ocular structure or adnexa. Examples may include uveitis (anterior, intermediate or posterior) dry eye, scleritis and conjunctival nodules among others. Keratic precipitates (KP) are cellular deposits found on the corneal endothelium, as seen on the anterior OCT images of this patient. The KP’s in this patient are large in size (>1mm diameter) so would be classified as mutton-fat KP and are typical of granulomatous uveitis. These large deposits typically contain macrophages and epithelioid cells. On further examination, grade 1 cells and flare were found so the patient was treated with corticosteroids and a cycloplegic agent. As ocular sarcoidosis can affect any of the ocular structures, a comprehensive full examination was also undertaken and a report sent to the patient’s treating physician.
CFEH Facebook Case #123
Case #123 PDF Download
For the original question click here
Answer
This presentation appears consistent with asymmetric posterior polymorphous dystrophy with the left eye more advanced than right. Posterior polymorphous dystrophy (PPMD) is a rare, autosomally dominant inherited condition with variable clinical expression between those affected. The characteristic signs of PPMD in this patient are the corneal vesicles (circular or oval transparent cysts with a gray halo) at the level of Descemet’s membrane. Other features of this condition can include band lesions or diffuse opacities. Band lesions range from 2 to 10 mm long and have parallel scalloped edges. The lesions may show shallow trenches and ridges on specular microscopy when there are a large number of confluent vesicles. Progression is variable in PPMD and patients may remain asymptomatic or the condition can progress causing corneal oedema and/or irido-corneal adhesions and peripheral anterior synechiae. These developments can cause visual disturbance and increased intraocular pressure, both of which require treatment.
CFEH Facebook Case #122
Case #122 PDF Download
For the original question click here
Answer
This patient has an epiretinal membrane (ERM) with associated vitreous adhesion and traction, causing distortion and thickening of the macular area, and an associated schisis. An ERM is thought to be formed when a partial posterior vitreous detachment results in residual vitreous material being left behind on the inner retinal surface. These residual cells proliferate to form an ERM which contracts causing macular pucker, and over time additional proliferation and contraction worsens the situation putting stress on the underlying retina. This effect is exacerbated when vitreous traction is also present. Vitreomacular traction (VMT) can cause similar macular changes (with or without an ERM) and this is an important differential in this case. VMT is defined by the literature as a partial posterior vitreous detachment that causes an anomalous distortion of the fovea (Dukker et al. 2013). The traction can cause macular pseudo-cysts, cystoid macular oedema and also macular schisis. Facebook case 122 It has been hypothesized that disruption of the foveal layers by ERM may pre-dispose towards the formation of idiopathic macular holes following PVD (natural or surgical – Bersirli et al. 2012). Additionally, ERMs can be associated with peripheral retinal degeneration including holes and tears. For that reason, detection of an ERM indicates the need for a dilated peripheral retinal examination. In this case, the patient has amsler distortion and the schisis is fairly extensive so referral to an ophthalmologist for assessment was recommended. Smaller, asymptomatic ERM’s may be managed with 6 monthly review to establish stability.
CFEH Facebook Case #121
Case #121 PDF Download
For the original question click here
Answer
This is a limbal dermoid (epibulbar dermoid) which is a benign, smooth white lesion typically found infero-temporally on the globe or limbus. They may be unilateral or bilateral and are believed to arise due to an embryonic anomaly rather than having a genetic cause. Limbal dermoids contain choristomatous tissue (tissue that is not normally found at that site). Limbal dermoids are usually graded according to the extent of tissue involvement: Grade I: superficial corneal involvement, less than 5mm diameter and localized to the limbus. Grade II: larger lesions covering most of the cornea, extending deep into the stroma but not involving Descemet’s membrane Grade III: involvement of the entire cornea and anterior chamber Grade I lesions usually show slow growth over time and cause oblique astigmatism as they flatten the adjacent cornea. This may cause amblyopia such as that seen in our patient. Interestingly, upon questioning our patient mentioned that she was born with only one kidney. This increases the likelihood that she has Goldenhar syndrome (oculo-ariculo-vertebral spectrum). This is a developmental malformation associated with limbal dermoids, auricular malformations and skeletal malformations. It typically affects one side of the body and additional manifestations can include congenital heart defects, renal defects (hypoplasia or failure of an organ to develop at all during embryonic growth), or central nervous system malformations (hydrocephalus, intracranial lipomas, cranial nerve dysgenesis).
CFEH Facebook Case #120
Case #120 PDF Download
For the original question click here
Answer
Optomap and Spectralis OCT images showed bilateral subretinal fluid, loss of photoreceptors and subretinal deposits with associated hyperautofluorescence. This presentation and early onset suggests Best’s disease (Best Vitelliform Macular Dystrophy), to be confirmed by genetic testing. Best’s disease is an autosomal dominantly inherited macular dystrophy characterized by bilateral multiple well-circumscribed yellow-orange macular sub-retinal lesions (vitelliform lesions). The disease onset is young (3-15 years of age) but diagnosis is typically delayed until the atrophic stage (after age 40) due to good visual acuity prior to this. Best’s disease usually diagnosed progresses through several stages: Progression through these stages is slow and variable, and may not correspond directly to visual acuity. Previtelliform stage: Normal macula/early RPE pigment changes. Acuity 6/6. Vitelliform stage: Well-defined, elevated, round lesion with an egg yolk appearance at the fovea. The rest of the fundus is normal and VA 6/6-6/15. Pseudohypopyon stage: Yellow material and fluid accumulates in the sub-retinal space. Can occur anywhere in the range of 8-38 years. Acuity is 6/6 – 6/15. Vitelloruptive stage: Vitelliform lesion breaks up causing a “Scrambled egg” appearance. Pigment changes and atrophy may be seen and there is a moderate reduction in acuity to 6/6 – 6/30. Atrophic stage: Re-absorption of the yellow material and atrophy of the RPE. Similar appearance to macular degeneration. Significant reduction in visual acuity to less than 6/60. CNVM/cicatricial stage: Choroidal neovascularisation may develop in response to retinal atrophy.
CFEH Facebook Case #119
Case #119 PDF Download
For the original question click here
Answer
This patient has bilateral central bull’s eye patterns of atrophy, mainly affecting the RPE and outer retinal layers, with associated VF loss (10-2 test grid). These changes correspond to a ring of hypofluorescence and surrounding mottling of mostly hyperfluorescence. These findings are consistent with a diagnosis of Hydroxychloroquine retinopathy. The risk factors for toxic retinopathy include: high dosage (>5.0mg/kg real weight), long duration of use (>5 years), renal disease, use of tamoxifen and concomitant macula disease (Marmor et. al. Recommendations on Screening for Chloroquine and Hydroxychloroquine Retinopathy 2016 Revision. Ophthalmol 2016; 123:1386-1394).
CFEH Facebook Case #118
Case #118 PDF Download
For the original question click here
Answer
This patient has moderate non-proliferative diabetic retinopathy (NPDR). There are multiple intra-retinal haemorrhages in both eyes, however these number less than what is required for severe NPDR (20 in each quadrant). OCT images show no sign of macular oedema. The Optometry Australia guidelines recommend a 3-6 month review for moderate diabetic retinopathy. Communication of the findings to the patient’s GP and endocrinologist is required. The ACCORD and FIELD studies have shown that fenofibrate may slow diabetic retinopathy progression in patients with mild to moderate NPDR at baseline. Both studies enrolled patients with type 2 diabetes who had, or were at high risk of having, cardiovascular disease.
CFEH Facebook Case #117
Case #117 PDF Download
For the original question click here
Answer
The term PED refers to a separation of the retinal pigment epithelium from Bruch’s membrane. PED’s may be caused by blood, serous fluid, drusen or a neovascular membrane and can be associated with a variety of conditions including age-related macular degeneration and pachychoroid spectrum disease. This patient had a serous PED (the PED is optically empty on OCT, indicating a likely serous nature). This resolved over a period of several years, leaving the macula apparently unaffected. This PED and the area of hypo-autofluoresence infero-nasal to the macula suggest a diagnosis of pachychoroid spectrum disorder, a condition characterized by increased choroidal thickness, areas of hyper and hypo autofluorescence that are in excess of RPE changes noted clinically, and the presence of small PEDs overlying areas of thickened choroid.
CFEH Facebook Case #116
Case #116 PDF Download
For the original question click here
Answer
Ocular Albinism. The anterior eye images show reduced pigmentation of both the iris and the lashes, most notable on the lower lid where eye makeup is absent. In the second image, retro-illumination defects of the iris may be clearly seen and the macular OCT scan shows an absence of the normal foveal pit and reflex (foveal hypoplasia). These signs, along with the reduced visual acuity and noted nystagmus are characteristic of ocular albinism. Other signs and symptoms that may be associated with ocular albinism include refractive errors, prominent choroidal vasculature, and optic disc hypoplasia. Ocular albinism usually shows an x-linked inheritance pattern, although occasionally can be autosomal recessive. Female carriers of the x-linked type can show partial iris translucency and scattered areas of depigmentation but are otherwise asymptomatic.
CFEH Facebook Case #115
Case #115 PDF Download
For the original question click here
Answer
The anterior OCT images show closing of the anterior chamber angle in the inferior region caused by the forward bowing of the inferior iris lesions. Examination with ultrasound biomicroscopy showed multiple cysts arising from the iris pigment epithelium from 5 to 7 o’clock in the right eye. The cysts are characterized by sonoluscent fluid within a thin and highly reflective wall. While primary iris pigment epithelial cysts are usually stationary, in this case the size and location of the cysts have given rise to significant bowing and obstruction of the inferior angle. Consequently regular monitoring was recommended for this patient, with gonioscopy to be performed annually.
CFEH Facebook Case #114
Case #114 PDF Download
For the original question click here
Answer
The slit lamp photo shows pigment dusting. The guttata are more obvious on the confoscan image while anterior OCT shows nodular formation of the endothelium and a thickened Descemet’s membrane. These clinical signs and the patient’s symptoms are consistent with a diagnosis of Fuch’s endothelial corneal dystrophy. This presentation is already relatively advanced. Fuch’s dystrophy typically appears in the 4th decade of life and is more common in females than males. Guttata usually start in the central cornea and over time expand to affect the peripheral cornea. The corneal endothelium has a “beaten metal” appearance and, as in this case, may show signs of pigment dusting. Over time, stromal oedema can progress to cause epithelial bullae or bullous keratopathy. Scarring, fibrosis and superficial vascularization of the cornea can occur secondary to the long-standing corneal oedema associated with this condition. In the early stages, hypertonic saline may be used to manage the corneal oedema, however as the disease progresses, surgical options must be considered. Due to the high rates of rejection and complications, penetrating keratoplasty is not the first choice of treatment for Fuch’s dystrophy. Newer techniques such as Descemet’s stripping automated endothelial keratoplasty (DSAEK) and Descemet’s membrane endothelial keratoplasty (DMEK) are preferred options. This patient was referred to a corneal specialist for consideration of surgery.
CFEH Facebook Case #113
Case #113 PDF Download
For the original question click here
Answer
OCTA works by repeatedly scanning an area of the retina and identifying changes in reflected B-Scan signals within a volume scan. The movement of erythrocytes through the blood vessels cause variations in the images which is interpreted as motion. In this way the technology allows us to visualize the retinal vasculature when there is blood flowing through the vessel. If there is no blood flow, or slow blood flow, motion is not detected and the vessel is then not “visible” on OCTA, such as in this case. While this technology is available commercially, the clinical understanding and applications are still evolving. The main advantages of this technology over traditional fluorescein angiography are that it is non-invasive (and can therefore be undertaken at every examination), it allows selective viewing of superior and deep retinal capillary plexus and choroidal vasculature, and it can potentially generate images with higher contrast and resolution than conventional fluorescein angiography. The limitations of this technology however mean that while it has great clinical application, it will never completely replace traditional FA. OCTA can’t show reduced perfusion or leakage, nor will it detect flow that is too fast or too slow. Additionally, the depth of penetration is limited and there is limited visualisation of small (less than 15μm) vessels. OCTA is best used in conjunction with other ocular imaging technologies and offers a non-invasive way to assess retinal vasculature however, the more invasive fluorescein angiogram will still be necessary in some patients.
CFEH Facebook Case #112
Case #112 PDF Download
For the original question click here
Answer
Optomap images show bone spicule intra-retinal pigmentation around the inferior arcades in both eyes while the superior retina appears unaffected. The retinal changes are clearly evident on fundus autofluorescence with large areas of the inferior retina showing hypo-fluorescence. There is a corresponding superior field defect in both eyes. This presentation is consistent with a diagnosis of sector retinitis pigmentosa. Sector retinitis pigmentosa (RP) is an atypical presentation of RP that typically involves the inferior retina. The affected area shows the characteristic bone-spicule pigmentation while the other retinal areas have a normal appearance. This condition is considered slowly progressive but given this patient’s excellent acuity and current age, the prognosis in this case is likely to be favourable. A binocular Esterman visual fields test was conducted to assess the patient’s vision for driving and the RP diagnosis was confirmed with electrophysiological testing.
CFEH Facebook Case #111
Case #111 PDF Download
For the original question click here
Answer
Collateral vessels can be seen on the retinal photos infero-temporally. These can also be seen on OCTA. Resolving dot and flame haemorrhages and microaneuryms can be seen in the photos, and macular oedema is also present. These findings are consistent with a history of branch retinal vein occlusion. Additionally, there are medium sized drusen temporal to the macula. This patient was referred to Ophthalmology for review.
CFEH Facebook Case #110
Case #110 PDF Download
For the original question click here
Answer
CHRPE with internal atrophy (lacunae) CHRPE are benign lesions, usually flat round and heavily pigmented. Approximately half of all CHRPE’s contain lacunae, or window-like defects of atrophy that are contained within the lesion such as in this case. These lacunae typically increase in size and also number over time, leading to a wide variety of different possible presentations. Some examples are seen below.
Routine review of these lesions are recommended. For more information on diagnosing retinal lesions, please see our pigmented and hypo-pigmented lesions chairside references, downloadable from our website.
)
CFEH Facebook Case #109
Case #109 PDF Download
For the original question click here
Answer
Vitreopapillary traction syndrome. This refers to the persistent attachment of the vitreous around the optic disc following a partial posterior vitreous detachment. Vitreopapillary traction with idiopathic ERM has been shown in the literature to be associated with altered architecture of the optic disc, increased average RNFL thickness, and visual field defects (Kim et al. 2014). Vitreopapillary traction can mimic disc oedema as it can cause blurring of the disc margins and obscuration of the vessels. OCT imaging is useful in differentiating this condition from true papilloedema. References Kim YW, Jeoung JW, Yu HG. Vitreopapillary traction in eyes with idiopathic epiretinal membrane: a spectral-domain optical coherence tomography study. Ophthalmology. 2014;121(10):1976-1982.PubMedGoogle ScholarCrossref 3. Houle E, Miller NR. Bilateral vitreopapillary traction demonstrated by optical coherence tomography mistaken for papilledema. Case Rep Ophthalmol Med. 2012;2012:682659.PubMedGoogle Scholar Simonett, J. Winges, K. Vitreopapillary Traction Detected by Optical Coherence Tomography JAMA Ophthalmol. 2018;136(5):e180727. doi:10.1001/jamaophthalmol.2018.0727
CFEH Facebook Case #108
Case #108 PDF Download
For the original question click here
Answer
Congenital optic disc pit. Congenital optic disc pits are hypo-pigmented oval or round excavations in the optic nerve head which can be bilateral in 10-15% of cases. Whilst typically seen temporally on the disc, they can be found at any location either centrally or at the edge of the disc. Acquired pits may be associated with high myopia or glaucoma. As neither of these are not present in this case, the pit is most likely congenital in nature. Optic disc pits can be associated with macular oedema, schisis or neurosensory detachment of the macula – termed optic disc pit maculopathy. Maculopathy will typically occur in the third to fourth decade of life, reducing acuity to 6/21 or worse. While spontaneous regression can occur, most cases have a poor prognosis with a gradual worsening of vision. The literature varies in estimating the prevalence of maculopathy but it is reported to be in the range of 25-75% of patients with optic disc pits.
CFEH Facebook Case #107
Case #107 PDF Download
For the original question click here
Answer
Pellucid marginal degeneration. This patient’s topography results show an inferior elevation of the anterior and posterior corneal surfaces with a crab-claw pattern of anterior corneal surface steepening. Corneal thicknesses at the pupil centre are within normal limits with the thinnest locations measuring 555μm. These findings are consistent with a diagnosis of pellucid marginal degeneration. Pellucid marginal degeneration is most common in males and is typically diagnosed in the 4th or 5th decade of life. It is associated with high against the rule astigmatism with near-normal visual acuity. Over time, PMD can progress, leading to more inferior corneal thinning and increased blur with spectacles. Management options include spectacles and/or RGP contact lenses with most cases able to be managed in this way. Very advanced cases can develop acute hydrops with corneal oedema and these cases may require surgical intervention in the form of penetrating keratoplasty, lamellar keratoplasty, INTACS or collagen cross-linking. For further information on differentially diagnosing corneal ectatic disorders, please refer to the CFEH Chairside reference “Corneal ectatic disease and thinning disorders”, available here
CFEH Facebook Case #106
Case #106 PDF Download
For the original question click here
Answer
Band keratopathy. The anterior eye photos showed a band-shaped, horizontal, grey corneal opacity within the intra-palpeberal fissure with involvement of the central cornea of the right eye. Cirrus anterior OCT imaging shows increased reflectivity in the Bowman’s layer in the affected area with associated posterior shadowing. These findings are consistent with a diagnosis of band keratopathy, which is a corneal degeneration characterised by calcium deposition mainly within Bowman’s layer. It may be associated with hypercalcaemia (among other causes), which in turn may be induced by systemic conditions such as vitamin D toxicity, hyperparathyroidism and sarcoidosis. This diagnosis should be communicated to the patient’s GP and investigations carried out for hypercalcaemia.
CFEH Facebook Case #105
Case #105 PDF Download
For the original question click here
Answer
This patient has nascent geographic atrophy. Nascent geographic atrophy is a term used to describe the ‘subsidence’ of the outer plexiform layer and inner nuclear layer without incomplete loss of the RPE and definite loss of photoreceptors. It can be found in an estimated 7% of eyes with intermediate AMD. Recently agreed nomenclature developed by the CAM (classification of macular atrophy) group identifies nascent geographic atrophy as a subset of iRORA (incomplete RPE and outer retinal atrophy), in AMD eyes without choroidal neovascularisation. In contrast, an OCT of a patient with geographic atrophy is below. It shows an absence of the RPE, inner segment ellipsoid zone, external limiting membrane and outer nuclear layer. Using the CAM group nomenclature, geographic atrophy is defined as a subset of cRORA (complete RPE and outer retinal atrophy).
The presence of nascent geographic atrophy is a prognostic sign seen in intermediate AMD that may herald imminent progression to geographic atrophy. A study by Wu et al. (2014) showed that on average, nascent geographic atrophy precedes the development of geographic atrophy by 11 months. For detailed information of this and other recently identified prognostic biomarkers for AMD, please refer to Angelica Ly’s open access review paper, available here.
)
CFEH Facebook Case #104
Case #104 PDF Download
For the original question click here
Answer
This is an advanced presentation of Stargardt Disease – a macular dystrophy caused by a mutation in the ABCA4 gene. It is characterised by progressive bilateral central vision loss, usually beginning in childhood or early adolescence. Stargardt disease is characterised by abnormal levels of lipofuscin and loss of the outer retinal layers. A reduction in lipofuscin density and RPE atrophy (as seen in the OCT image) have caused the hypo-autofluoresence noted in this patient. In earlier presentations when this condition is typically diagnosed, you would expect to see yellow-white retinal flecks which show a mixture of hyper and hypo-autofluorescence as in the images below.
)
CFEH Facebook Case #103
Case #103 PDF Download
For the original question click here
Answer
This first OCT Image show a paravascular cyst associated with a vascular microfold. The cyst is defined as a small acoustically empty space adjacent to a retinal vessel. These may be found with OCT in up to 50% of high myopes and are incidence increases with age, axial length, degree of myopia and presence of posterior staphyloma. Rupture of these cysts causes a paravascular lamellar hole which may be seen in the second OCT image inferior to the retinal cyst. Paravascular lamellar holes are thought to have an incidence of up to 30% and are most frequently found along the inferior vascular arcades and at the edge of peripapillary staphylomas. Paravascular lamellar holes that form over areas of chorioretinal atrophy have been associated with rhegmatogenous retinal detachment.
CFEH Facebook Case #102
Case #102 PDF Download
For the original question click here
Answer
Morning glory anomaly. This anomaly is congenital and typically unilateral in presentation. It is characterised by a large funnel-shaped excavation of the disc, usually with an associated white glial tuft in the centre of the disc. This tuft is caused by a persistent hyaloid vascular remanent – seen in this case at the centre of the excavated disc in the middle OCT image. The disc is typically orange/pink in colour with surrounding chorioretinal pigment changes in a ring-shaped pattern and the blood vessels are increased in number, emerging from the excavated disc in a radial pattern rather than in a typical branching pattern. From the OCT images, we can also see paravascular cysts superior and inferior to the disc with associated vitreo-retinal traction. Inferiorly there is also a mild retinoschisis present. The B-scan ultrasound shows a posterior staphyloma and the morning glory disc is evident. Morning glory anomaly has a strong association with serous retinal detachment the incidence of which is commonly reported at 30%. Presentation may be sporadic, or have systemic associations such as frontonasal dysplasia (mid-facial anomalies) and midline brain malformations.
CFEH Facebook Case #101
Case #101 PDF Download
For the original question click here
Answer
An 52 year old Asian male presented to CFEH for a macular review. In 2016 vitreomacular adhesion (VMA) was noted in his left eye. VMA is defined as adhesion of the vitreous to the macular with no associated change to the foveal contour or underlying retinal tissue. At the 12 month review, this VMA has progressed to vitreomacular traction (VMT) with distortion of the foveal pit and intra-retinal anomalies. For a more in-depth exploration of vitreo-retinal interactions, join Lead Clinician Paula Katalinic and staff optometrist Henrietta Wang for their webinar presentation “Posterior Vitreous Detachment – Rules of Traction” next Tuesday evening at 6:30pm AEST (see www.learningforvision.com.au for details). A free chairside reference covering the diagnosis and management of vitreomacular conditions can be downloaded from the CFEH website by clicking here.
)
CFEH Facebook Case #100
Case #100 PDF Download
For the original question click here
Answer
An 89 year old European male was seen at CFEH as part of a study looking at the triaging of referrals to a local hospital ophthalmology department. This study is part of research being conducted by the Centre into alternate models of collaborative care. His best corrected acuity in this eye was 6/9.5. Based on the information supplied in the referral letter, this was categorized as a non-urgent referral. Imaging conducted at the Centre revealed late (exudative) AMD which substantially changed the management of this patient. Arrangements were subsequently made for a prompt assessment at the hospital eye clinic. This case illustrates the need for novel clinical pathways to ensure timely diagnosis of vision threatening eye conditions such as exudative AMD, proliferative diabetic retinopathy and disorders of the visual pathway.
)
CFEH Facebook Case #99
Case #99 PDF Download
For the original question click here
Answer
The anterior OCT and photos show stromal haze in both eyes, affecting the paracentral cornea in the right eye and the central cornea in the left. Guttata were bilaterally on slit lamp and the OCT images show a thickened epithelial layer. Confoscan endothelial cell count results show a cell count of 425 cells/mm2 in the right eye and 1440 cells/mm2 in the left. Both eyes are significantly below the “normal” range for this patient’s age of 1751-3279 cells/mm2. The confocan images also highlight the guttata (appearing as dark areas in the magnified images, most notable in the right eye). Given the stromal haze, guttata, reduced endothelial cell counts and reduced vision, this patient was recommended referred to a corneal specialist. As the patient has had a corneal graft, these signs may be indicators that the graft is failing particularly given his recent symptoms of reduced vision.
CFEH Facebook Case #98
Case #98 PDF Download
For the original question click here
Answer
This patient has a slight silvery glow around the inferior venous arcade, seen on Optomap. There is a corresponding hyper-autofluorescence. These findings are consistent with a previous periphlebitis. This is an inactive presentation of the condition – active periphlebitis typically presents with localised inflammatory infiltrates surrounding the retinal veins. Multiple sclerosis causes the degeneration of axons and approximately 10% of sufferers show retinal periphlebitis. Recent studies have suggested a correlation between the presence of periphlebitis and increased disease activity and suggested this as a prognostic biomarker for disease severity (Ortiz-Perez et al. 2013). Increased disease activity is significant as it is associated with more severe brain atrophy and disability (Barkhof et al. 2009). Other possible causes of periphlebitis include sarcoidosis, lyme disease, pars planitis, tuberculosis and Eales disease. References Barkhof F, Calabresi PA, Miller DH, Reingold SC. Imaging outcomes for neuroprotection and repair in multiple sclerosis trials. Nat Rev Neurol 2009;5:256–266 Ocular pathology in multiple sclerosis: retinal atrophy and inflammation irrespective of disease duration Ari J. Green,corresponding author1 Stephen McQuaid,3 Stephen L. Hauser,1 Ingrid V. Allen,2 and Roy Lyness Ortiz-Pérez, S. Martínez-Lapiscina, E. Gabilondo, I., Fraga-Pumar, E., Martínez-Heras, E., Saiz, A., Sanchez-Dalmau, B., Villoslada, P. (2013) Retinal periphlebitis is associated with multiple sclerosis severity. Neurology. 2013 Sep 3; 81(10): 877–881.
CFEH Facebook Case #97
Case #97 PDF Download
For the original question click here
Answer
Imaging shows a lesion temporal to the macula with associated retinal striations and traction. This traction has caused an ILM detachment and the formation of cystic spaces in the inner nuclear layer (as seen on OCT). These findings are consistent with a diagnosis of combined hamartoma of the retinal and RPE (CHRRPE) which is causing retinal traction in the left eye. CHRRPE are benign elevated tumours and the level of the sensory retina and RPE. Pigmentation can be variable and the lesions are often associated with epiretinal membrane formation (as in this case here). Lesions can cause significant visual disturbance in some cases, depending on their location. They may also progress with time causing complications such as vitreous haemorrhage, neovascularization, macular hole, and peripheral hole formation. Recently Dedania et al. (2018) devised a classification system for CHRRPE which takes into account the location of the lesion, the retinal layers affected and the level of traction noted. Based on the classification derived, they also developed recommendations for follow up intervals ranging from 2 to 12 months. The literature does report an association with Neurofibromatosis 1 and 2 and a bilateral presentation of CHRRPE should increase suspicion of these conditions. Reference Dedania, V., Ozgonul, C., Zacks, D., Besirli, C. (2018) Novel classification system for combined hamartoma of the retina and the retinal pigment epithelium. Retina 38:12–19, 2018
CFEH Facebook Case #96
Case #96 PDF Download
For the original question click here
Answer
This patient has a retinal embolism, most likely a Hollenhorst plaque. The American Heart Association guidelines (2009) recommend that patients with transient ischaemic attack (TIA) or transient retinal ischaemia undergo a full cardiovascular work-up to determine the aetiology within 24 hours of the onset of symptoms. This is based on studies which show these patients have a 10% risk of stroke within 90 days (it is worth noting that half of these occur within the first 48 hours). Retinal artery occlusions have a high association with subsequent stroke and stroke-related mortality – calculated at nearly 3 times higher than those without retinal emboli at baseline in a study by Wang et al (2006). Asymptomatic retinal emboli is associated with a moderately increased risk of associated mortality (independent of age, sex and vascular risk factors). This patient was advised to undergo a full cardiovascular work-up within 1-2 days. References 1. Wang et al. (2006) Retinal Arteriolar Emboli and Long-Term Mortalilty – Pooled Data Analysis From Two Older Populations. Pub Stroke 2006;37:1833-1836. 2. Vodopivec et al. (2017) Management of Transient Monocular Vision Loss and Retinal Artery Occlusions. Pub Seminars in Ophthalmology 2017; 32(1) 125-133
CFEH Facebook Case #95
Case #95 PDF Download
For the original question click here
Answer
The disc photos show horizontally oval, large sized discs with large cups and extensive circumferential peripapillary atrophy. There were no signs of Drance haemorrhage or notching. The OCT results reveal signs of pathological myopia including posterior staphyloma, localised peripapillary detachment, peripapillary schisis, and paravascular cysts. The visual field results show enlarged blind spots with deep depressions secondary to these pathological changes. To sumarise, there is no convincing evidence of glaucoma with the field defect consistent with pathological myopia.
CFEH Facebook Case #94
Case #94 PDF Download
For the original question click here
Answer
Vortex vein varix. OCT imaging allows us to scan through the elevation and measure its maximum height which is found to be just under 800μm. The OCT images show an optically empty thickening of the choroid with significant dilation of the choroidal veins. There is a subtle posterior displacement of the sclera and no changes to the overlying retinal architecture. This presentation is consistent with a diagnosis of vortex vein varix, which represents a physiological dilation of one of the choroidal vortex veins. This is a benign condition that can sometimes mimic a choroidal melanoma. During an examination, the two can be distinguished most easily by observation with a binocular indirect ophthalmoscope. A vortex vein varix will typically flatten or collapse in certain directions of gaze and may be flattened with manual scleral depression while a melanoma, being a solid lesion, will not.
CFEH Facebook Case #93
Case #93 PDF Download
For the original question click here
Answer
A 65 year old Asian male presented for examination. Below are the Optomap and Fundus Autofluorescence images for his left eye. This case is a good illustration of the value of different imaging modalities. The Optomap image clearly shows asteroid hyalosis while the fundus autofluorescence image highlights the grouped CHRPE (bear tracks). CFEH provides advanced eye imaging and visual system diagnostic services with no expense to patients for both anterior and posterior eye conditions. To register with us or refer a patient, please click the link: https://centreforeyehealth.com.au/publications/
)
CFEH Facebook Case #92
Case #92 PDF Download
For the original question click here
Answer
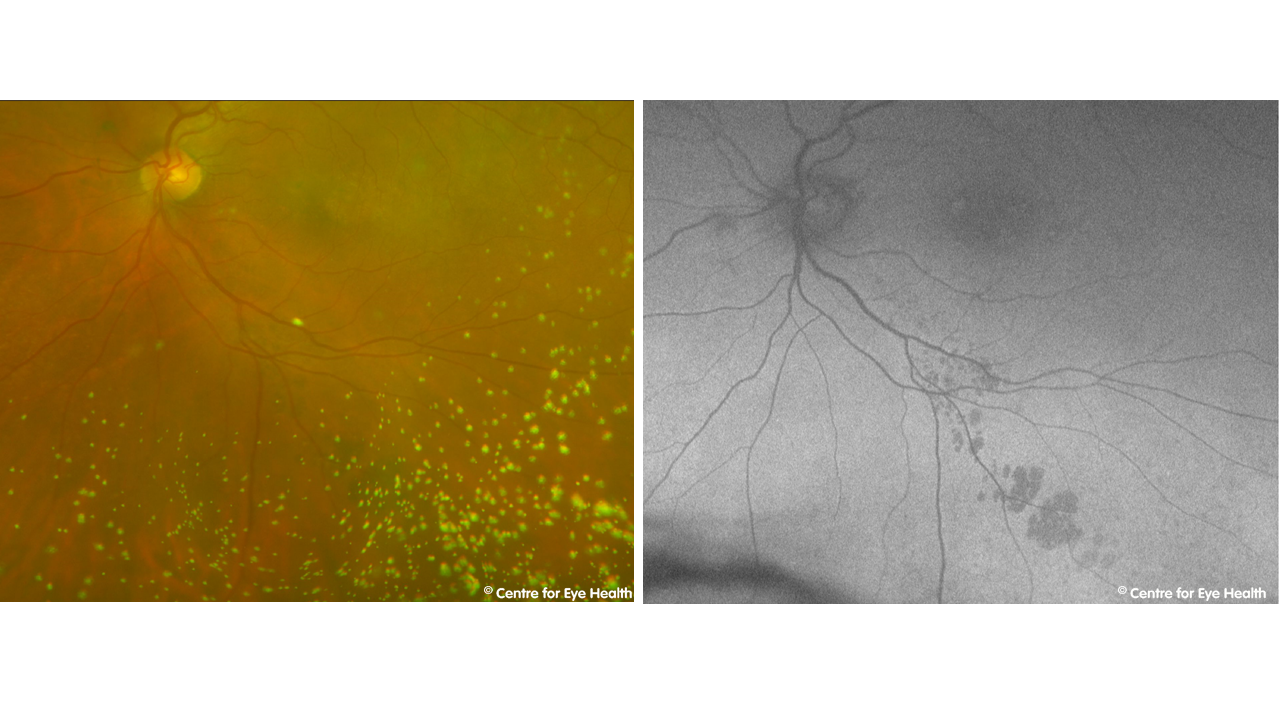
Confluent paving-stone degeneration. Paving-stone degeneration is a term referring to areas of chorio-retinal atrophy with discrete margins, located between the ora serrata and the equator. It is characterised by an atrophy of the outer retinal layers and adherence of the inner retinal layers to Bruch’s membrane. Lesions are typically yellow-white in colour due to choroidal atrophy (causes the sclera to be partially visible). Paving-stone degeneration can become confluent over time, as in the case presented here. The discrete lesions can still appreciated on the fundus autofluorescence image, but are more difficult to discern on the Optomap image. A different presentation of extensive paving-stone degeneration is below:
Paving-stone degeneration is a benign condition, not associated with any known complications.
)
CFEH Facebook Case #91
Case #91 PDF Download
For the original question click here
Answer
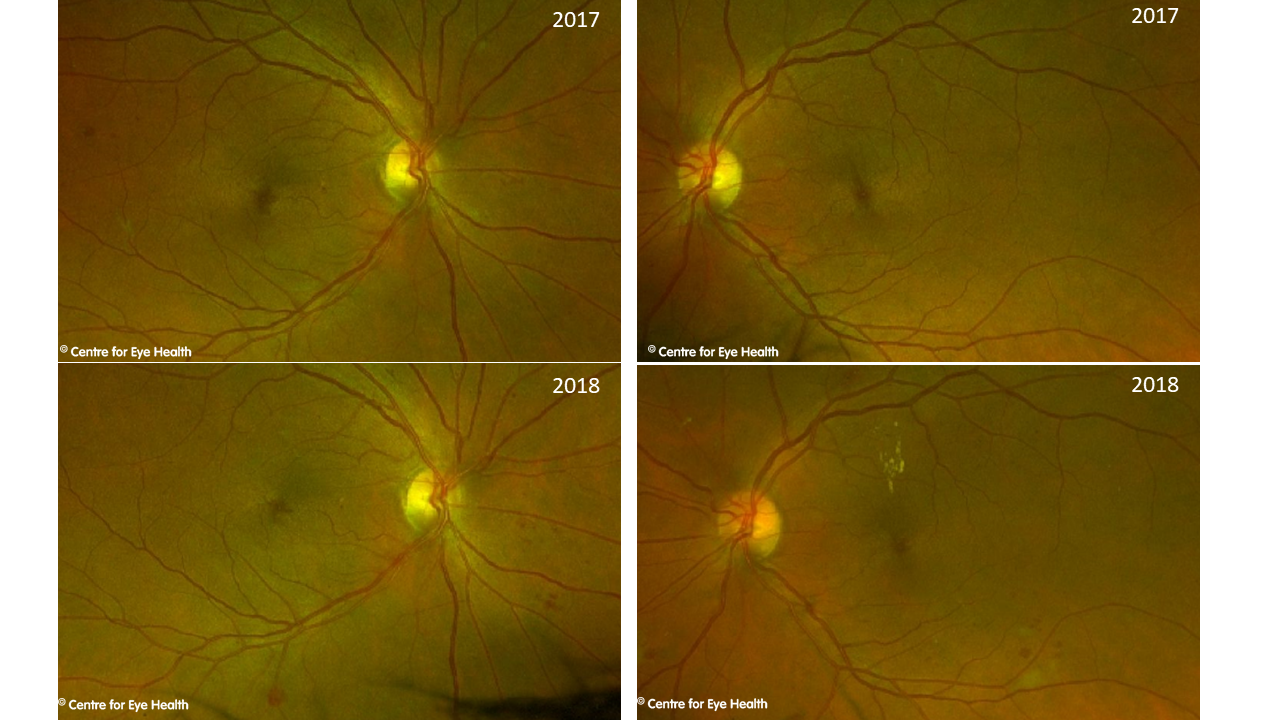
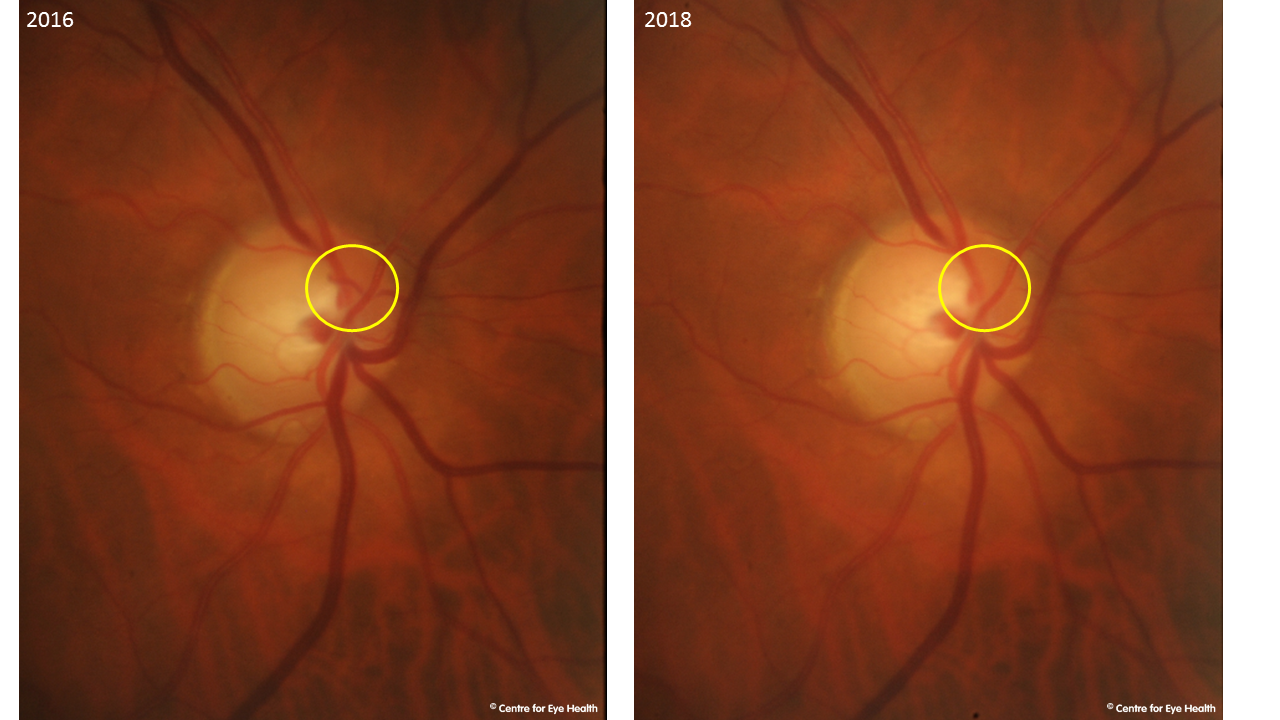
There is marked attenuation of a small venule overlying the superonasal disc as compared to the 2016 baseline such that the vessel is no longer visible (see images). These vessel changes suggest hypertensive retinopathy and may possibly indicate an impending vein occlusion. There was no discernable change to the neuroretinal rim when stereoscopic comparison was made using flicker comparison. The vascular changes are likely related to the elevated blood pressure and this patient was instructed to see his GP as soon as possible for reassessment of his blood pressure and appropriate management.
)
CFEH Facebook Case #90
Case #90 PDF Download
For the original question click here
Answer
The darker area at 3 o’clock is an oral bay and the white dot-like lesion an oral pearl. An oral bay is formed at the ora serrata when 2 or more ora “teeth” (or dentate processes) join. These may appear similar to a retinal hole, however oral bays do not increase the risk of retinal detachment. Oral pearls are drusen-like deposits between bruch’s membrane and the retinal pigment epithelium which appear as glistening white deposits. They are usually located at the ora “teeth” or less commonly in an oral bay, as in our patient. There are also extensive areas of white without pressure visible in the temporal periphery of this patient.
CFEH Facebook Case #89
Case #89 PDF Download
For the original question click here
Answer
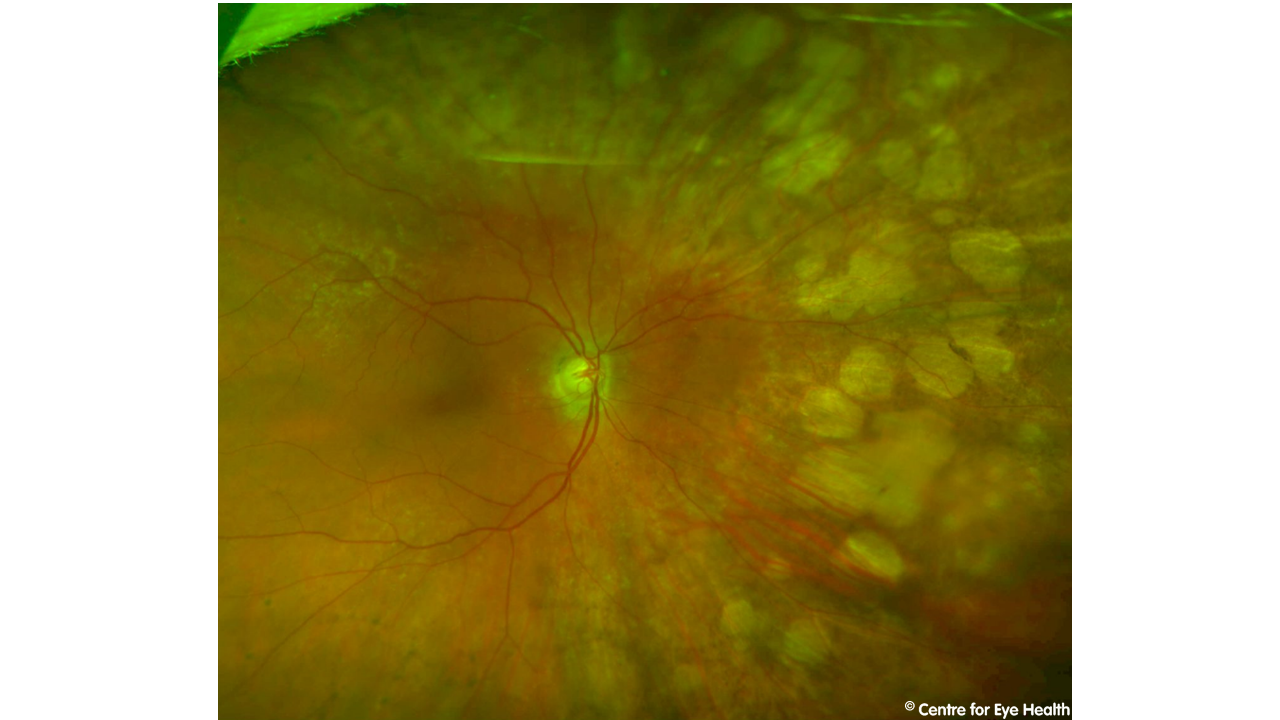
Retinal images show a patchy bilateral retinal atrophy around the maculae with a ring-shaped appearance and a small central region of relative sparing. OCT shows a distortion of the foveal pit and disorganisation of the retinal layers. There is loss of the outer retinal layers (the outer nuclear layer, inner segment ellipsoid layer, external limiting membrane and RPE). There is also a hyper-reflective sub-retinal elevation with posterior shadowing. These findings are consistent with a diagnosis of either central areolar choroidal dystrophy (CACD), although a diagnosis of cone dystrophy must also be excluded. CACD is an inherited condition characterised by an area of photoreceptor, RPE and choriocapillaris atrophy at the central macula. Patients typically present with reduced vision in their 40-50’s. Progressive atrophy eventually affects the fovea around the age of 70, causing significant central vision loss. Clinically, OCT shows loss of the outer retinal layers, such as in this patient and a ring of hyper-autofluorescence in early stages, followed by an area of marked hypo-autofluoresence in the later stages. Electrophysiology testing (below) confirmed a diagnosis of macular dystrophy. For further information on the clinical uses and basic interpretation of electrophysiology you can register here to attend our February webinar “Electrophysiology 101 – what every optometrist needs to know”, presented by Michael Yapp and Dr Nagi Assaad on Tuesday 13th February 2018 at 6:30pm.
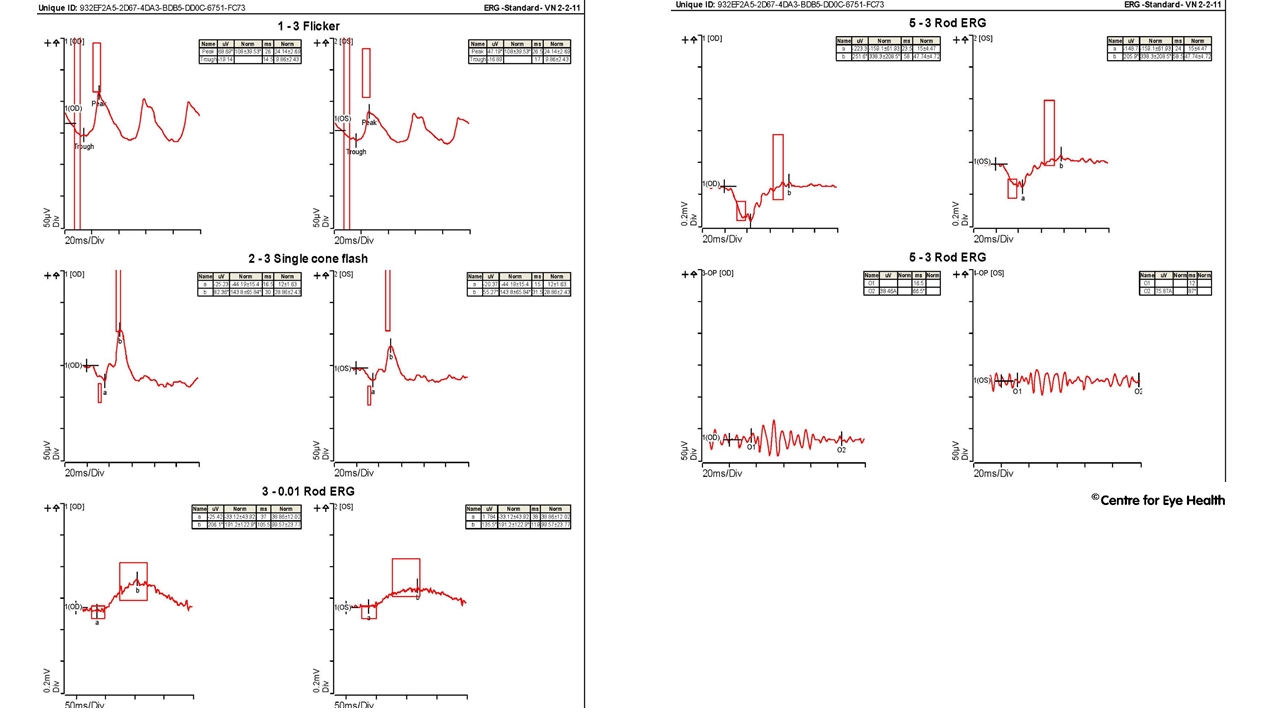)
CFEH Facebook Case #88
Case #88 PDF Download
For the original question click here
Answer
There is temporal disc pallor in the right eye with an associated wedge defect encompassing the papillomacular bundle and an absent macular reflex in this eye. The GCA also show marked loss of the inner retinal layers in this region. These clinical findings are consistent with an old occlusion of the cilioretinal artery. Additional findings include druplets at the right macula and generalised vascular tortuosity with copper wiring and arteriovenous nipping. This patient was referred to an Ophthalmologist for further assessment.
CFEH Facebook Case #87
Case #87 PDF Download
For the original question click here
Answer
There is a large cotton wool spot visible just superior to the optic nerve which is imaged on OCT (far left of the image) and a blot haemorrhage adjacent to the superior arcades (not visible in the photos). Additionally, there were collateral vessels at the nasal neuro-retinal rim and supero-temporal to the disc. The clinical presentation for this patient does not indicate a diagnosis of glaucoma. The vascular changes noted are likely to be related to a previous venous occlusion. This patient was referred to her GP for a systemic vascular workup.
CFEH Facebook Case #86
Case #86 PDF Download
For the original question click here
Answer
Retinopathy of prematurity. This patient was born prematurely at 30 weeks gestation. The Optomap and fundus autofluorescence images show vascular sheathing at the disc and peripheral retina in both eyes, tortuous blood vessels, peripheral retinal hyperplasia (more notable in inferiorly in the right eye and temporally in the left), circumferential retinoschisis with vitreomacular traction in the left eye and an infero-temporal ridge in the right eye. The optic disc also showed signs of dragging away from the fovea in the right eye. Retinopathy of prematurity has 2 main phases – firstly there is delayed growth of the retinal vessels after birth, and a partial regression of vessels that have developed. This is followed by the growth of pathological vessels, stimulated by retinal hypoxia. The main risk factors for this condition include preamture birth (before 31 weeks), a low birth weight (less than 1.25kg) and the use of oxygen following birth. The smaller the baby and earlier the birth, the higher the risk of ROP, however not all premature babies will develop this condition. The pathophysiological process is, in brief, as follows. The high oxygen levels suppress VEGF in the early stages of ROP, inhibiting normal vessel growth. This is followed by the second phase of ROP where retinal hypoxia induces high levels of VEGF caucsing pathological retinal vessels to develop. Tractional retinal detachment(s) can occur in the later stages of this disease. This condition is classified into 5 stages, ranging from stage 1 (mildly abnormal blood vessel growth requring no treatment) through to stage 5 (tractional retinal detachment and blindness). In severe cases, timely treatment in the form of laser therapy or cryotherapy is required to avoid vision loss.
CFEH Facebook Case #85
Case #85 PDF Download
For the original question click here
Answer
Multiple Evanescent White Dot Syndrome. The fundus autofluoresence image shows multiple hyperfluorescent lesions at the posterior pole, particularly around the paramacular region, and extending to the nasal periphery. An OCT line scan through the macula shows small multiple outer retinal disruptions at the ellipsoid level and foveal granularity. This presentation is consistent with a diagnosis of MEWDS. This is one of a group of retinal conditions termed “white dot syndromes” and is an idiopathic, spontaneously resolving inflammatory disorder typically affecting the 20 to 45 year age group with a strong female predilection. Symptoms can include a sudden reduction in visual acuity, photopsia, temporal or paracentral scotomatas, and dyschromatopsia. The condition is usually unilateral, as in this case, however bilateral cases have been reported. MEWDS has a good prognosis with recovery of the Ise line typically occurring over 4.5-6 weeks with concurrent resolution of the hyperautofluresence. Rarely, some patients may have a persistent blind spot enlargement, photopsias, and dyschromatopsia. For further information about this and other white dot syndromes, please refer to the CFEH paper ” OCT and Fundus Autofluorescence Enhances Visualization of White Dot Syndromes” which can be downloaded using the following link: https://www.ncbi.nlm.nih.gov/pubmed/25875689
CFEH Facebook Case #84
Case #84 PDF Download
For the original question click here
Answer
Peripapillary pachychoroid syndrome. Pachychoroid spectrum disorder is a term referring to a group of disorders with the common features of increased choroidal thickness, reduced fundus tessellation, drusenoid RPE changes, areas of hyper and hypo autofluorescence that are in excess of RPE changes and the presence of small PEDs overlying areas of thickened choroid. The conditions that are generally accepted to fall under this umbrella include central serous chorioretinopathy (CSCR), pachychoroid epitheliopathy (PPE – Facebook case 55), polypoidal choroidal vasculopathy (PCV- Facebook case 80) and pachychoroid neovasculopathy. A 2017 article by Phasukkijwatana et al (RETINA 0:1–16, 2017) however introduced another variant within this spectrum – peripapillary pachychoroid syndrome. The features of this condition can be seen in our patient. Peripapillary pachychoroid syndrome has been identified as including pachychoroid features surrounding the optic nerve, associated with intraretinal or subretinal fluid and sometimes optic nerve head oedema. The presence of pachyvessels and serous PED’s (pigment epithelial detachments) are associated with this condition. The EDI-OCT images of our patient show these features in the peripapillary area, including pachyvessels:
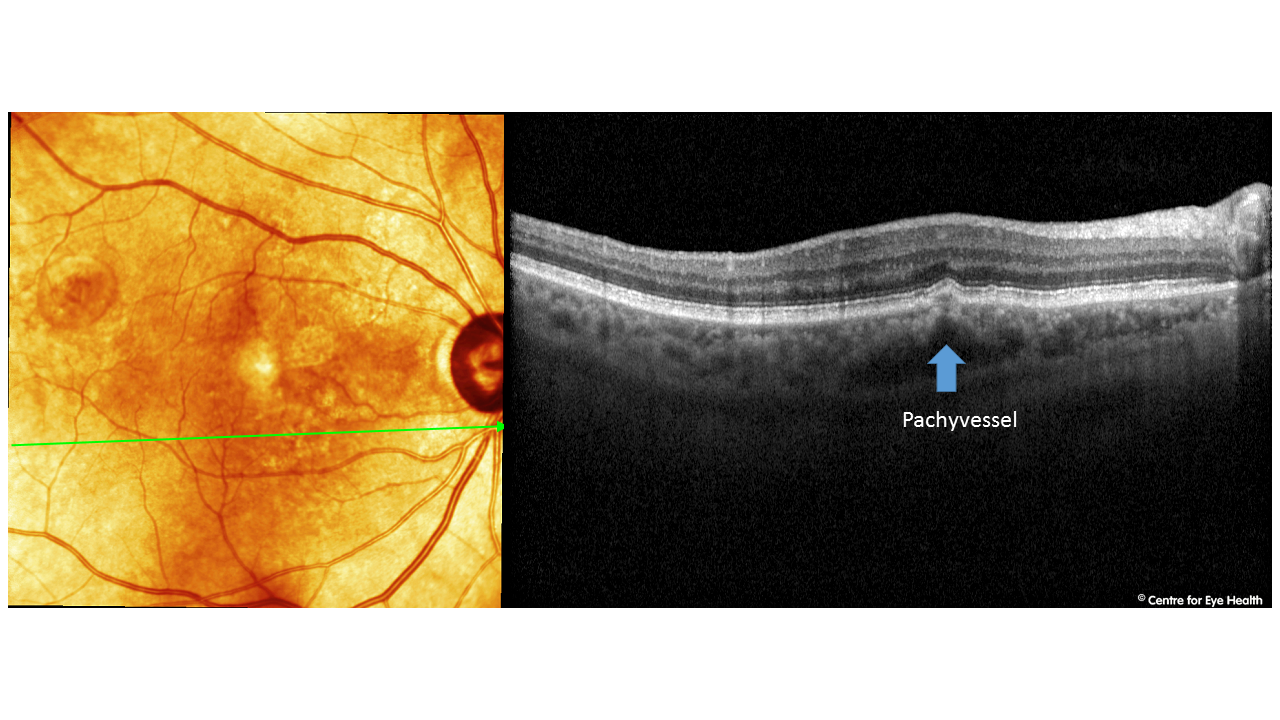)
CFEH Facebook Case #83
Case #83 PDF Download
For the original question click here
Answer
OCT imaging shows the presence of sub-retinal fluid and OCTA shows anomalous vessels in the choriocapillaris, consistent with a diagnosis of choroidal neovascularisation. This patient was referred to Ophthalmology for suspected exudative AMD. This case is a good illustration of the additional information that can be obtained using advanced imaging. It has also been shown through a recently published CFEH study that specialised training in the use and interpretation of advanced imaging is essential to maximise this advantage and improve the stratification of AMD. For the full paper, please click on this link: http://onlinelibrary.wiley.com/doi/10.1111/cxo.12607/full
CFEH Facebook Case #82
Case #82 PDF Download
For the original question click here
Answer
It is an area of lattice degeneration with marked vitreoretinal traction. There is an associated retinoschisis and tractional detachment. The true nature of this lesion can only be appreciated with OCT imaging: This patient was referred promptly to an ophthalmologist. A useful chairside reference on peripheral retinal lesions is available to download at: https://www.centreforeyehealth.com.au/wp-content/uploads/2019/01/chairside_reference_peripheral-retina.pdf
)
CFEH Facebook Case #81
Case #81 PDF Download
For the original question click here
Answer
It is an area of dark without pressure. Dark without pressure refers to an area of darker retina with well-defined edges that are often scalloped. The OCT image through the lesion shows a change in the reflectivity of the ellipsoid zone in the area of the lesion. These lesions are benign but can change in appearance over time. For a handy guide to differentially diagnosing peripheral retinal lesions, download the CFEH Chairside reference at: https://centreforeyehealth.com.au/wp-content/uploads/2016/09/chairside_reference_peripheral-retina.pdf
CFEH Facebook Case #80
Case #80 PDF Download
For the original question click here
Answer
This patient polypoidal choroidal vasculopathy in the left eye (PCV). The retinal photo shows significant subretinal fibrosis temporal to the optic disc and involving the centre of the macula. OCT analysis shows loss of the outer retinal layers in this area and a serous sub-RPE elevation nasally with several smaller PEDs temporally. Superior to the macula there are several highly peaked PEDs, some of which may be haemorrhagic in nature, and there is sub-retinal fluid present, suggesting an active presentation of the disease. The first OCT image has a “double layer sign” – a highly reflective layer underneath the RPE. The OCT underneath this indicates the presence of a polyp. The incidence and demographic features of PCV varies among different ethnic groups. The incidence of PCV is high in African Americans, moderately high in Asians, and relatively low in Caucasians (Sho, Takahashi et al. 2003). In African Americans, PCV affects middle-aged women more frequently and the lesions are usually bilateral – occurring in the peripapillary region (Yannuzzi, Wong et al. 1999; Ciardella, Donsoff et al. 2004). In Asian patients, PCV tends to be more common in males as a unilateral condition affecting the macula (Kwok, Lai et al. 2002; Anantharaman, Ramkumar et al. 2010). In Caucasians, the disease is more commonly a bilateral condition with peripapillary lesions and no (Yannuzzi, Wong et al. 1999) or female (Yannuzzi, Ciardella et al. 1997) gender predilection. It presents earlier in life than neovascular AMD. In Asian populations it has been found that between 20% and 50% of macular exudation and haemorrhage are found to have PCV. On fundus imaging, neovascular AMD and PCV are impossible to differentiate. OCT gives more insight with PCV complexes causing focal highly peaked, u-shaped elevations of the RPE and an appearance typical of polyps. Associated serous retinal detachments are usually seen, as in this case. Compared with neovascular AMD, PCV usually shows more serous retinal detachments and less intraretinal oedema. Characteristic of this condition however is the “double-layer sign” on OCT such as seen in OCT image 1. For more information on PCV and the OCT presentations of this disease, please join our November 14th webinar on the topic “Fundus changes in Choroidal Disease”. For further information, please contact learningforvision@cfeh.com.au.
CFEH Facebook Case #79
Case #79 PDF Download
For the original question click here
Answer
This patient has gyrate atrophy. Retinal imaging shows multiple areas of chorioretinal atrophy surrounding the posterior pole and midperipheral retina. There are well-defined areas of hypo-autofluorescence on fundus autofluorescence scans, with hyper-autofluorescence seen at the maculae. Spectralis OCT line scans through areas of atrophy revealed thinning of the outer retinal layers and RPE. Imaging of the macula revealed macular thickening, outer retinal tubulation and mild cystoid macular oedema at the edge of atrophic areas near the fovea. Gyrate atrophy is caused by a mutation to the ornithine aminotransferase (OAT) gene, resulting in a reduced amount of functional OAT enzyme. As a result, there is an accumulation of plasma ornithine which is toxic to the RPE and choroid. Early in the disease process, patients develop large peripheral areas of chorioretinal atrophy. These areas eventually coalesce forming a scalloped border peripherally, at the junction of normal and abnormal retina. Nyctalopia (night blindness) usually occurs in the first decade of life, followed by a progressive loss of visual field and eventually central vision is also lost. Treatment may involve dietary restriction of ornithine however success of this is limited. A small percentage of those with this condition do respond to vitamin B6 supplementation and this lowers the plasma ornithine levels however for most there is no effective treatment.
CFEH Facebook Case #78
Case #78 PDF Download
For the original question click here
Answer
This patient has pseudo-papilloedema due to small crowded discs. She does not require referral. The disc photos show no visible cup and indistinct margins which are due, in this case, to an average number of axons trying to fit through a small scleral foramen and converging at a small optic disc. Spectralis OCT shows obliquely inserted small, crowded discs with no signs of disc drusen. There are some hypo-reflective cystic spaces in the inner nuclear layer and outer nuclear layer at 4o’clock of the disc margin. These are likely due to optic nerve protrusion leading to separation and stretching of the retinal layers. Fundus autofluorescence is unremarkable and shows no hyper-autofluorescence that might indicate optic nerve head drusen.
CFEH Facebook Case #77
Case #77 PDF Download
For the original question click here
Answer
The large epiretinal membrane seen on Optomap and associated retinal traction have caused the formation of a macular pseudohole. The appearance on Optomap is similar to that of a full thickness macular hole, however OCT imaging shows it to be a distortion of the foveal pit secondary to traction from the epiretinal membrane. A useful optometric test to differentiate full thickness macular holes from pseudoholes is the Watzke-Allen test whereby a slit lamp beam is focused on the macula. If the beam appears broken, it is likely the patient has a true macular hole while if it is unbroken, it is likely to be a pseudo-hole. This test has been shown to have extremely high sensitivity and specificity.
The OCT findings are below:
)
CFEH Facebook Case #76
Case #76 PDF Download
For the original question click here
Answer
The clinical findings are consistent with a diagnosis of fundus flavimaculatus. The fleck-like yellow lesions surrounding both maculae spare the fovea and correspond to areas of mixed stipled hyper and hypo-autofluorescence. OCT imaging shows the lesions to be causing disruption to the RPE and ISe line, with some focal areas of subretinal/ RPE deposits. Fundus flaviamaculatus is part of the same disease spectrum as Stargardt’s disease but has a later age of onset and slower progression. Both are related to a defect of the ABCA4 gene. The flecks seen in fundus flavimaculatus can take many different shapes, including round, pisciform, butterfly or spear-like. They initially appear yellow-white in colour and are well defined, although over time they change in appearance becoming grey, fuzzy and ill-defined. The flecks may also be found at variable levels of the retina although they are typically at the level of the RPE (type 1) or the outer nuclear layer (type 2).
CFEH Facebook Case #75
Case #75 PDF Download
For the original question click here
Answer
Solitary idiopathic choroiditis. The lesion is a yellow-white and discrete in nature with a surrounding orange halo – an appearance that is consistent with a diagnosis of solitary idiopathic choroiditis. OCT imaging confirms the diagnosis as the lesion can clearly be seen coming up from the sclera and compressing the overlying choroid and choriocapillaris. The lesion is inactive as there is no sign of yellow intraretinal exudation, localized subretinal fluid or focal retinal hemorrhages and the border of the lesion is well defined. For further information on differentially diagnosing lesions of the posterior eye, please see the recently released CFEH Chairside references “Hypo-pigmented lesions of the posterior eye” and “Pigmented lesions of the posterior eye” which may be downloaded for free from the CFEH website by clicking on the links above.
CFEH Facebook Case #74
Case #74 PDF Download
For the original question click here
Answer
The patient was referred to an ophthalmologist who elected to monitor the serous pigment epithelial detachment (PED) for the development of choroidal neovascularisation. When he was referred back to the Centre for follow up scans after 6 months, the PED had almost completely resolved leaving little sign that it had ever been there aside from a mild thinning of the RPE layer, slight loss of reflectivity of the ISe line and subtle irregularities in the spectral reflectance map (left).
)
CFEH Facebook Case #73
Case #73 PDF Download
For the original question click here
Answer
The discs are very difficult to assess due to a combination of the inherent tilting, marked PPA and underlying peripapillary intra-choroidal cavitation. The OCT RNFL analysis also needs to be viewed with caution due to variable segmentation associated with the same factors and the GCA analysis was unable to segment the layers accurately. Below are the visual fields for this patient: There is a notable general depression in the right compared to the normative data with the pattern deviation showing a significant superior arcuate visual field defect and a suspicious nasal depression in the left. Based on the clinical picture a whole and in conjunction with a glaucoma specialist, this patient has been determined to have glaucoma and is being treated.
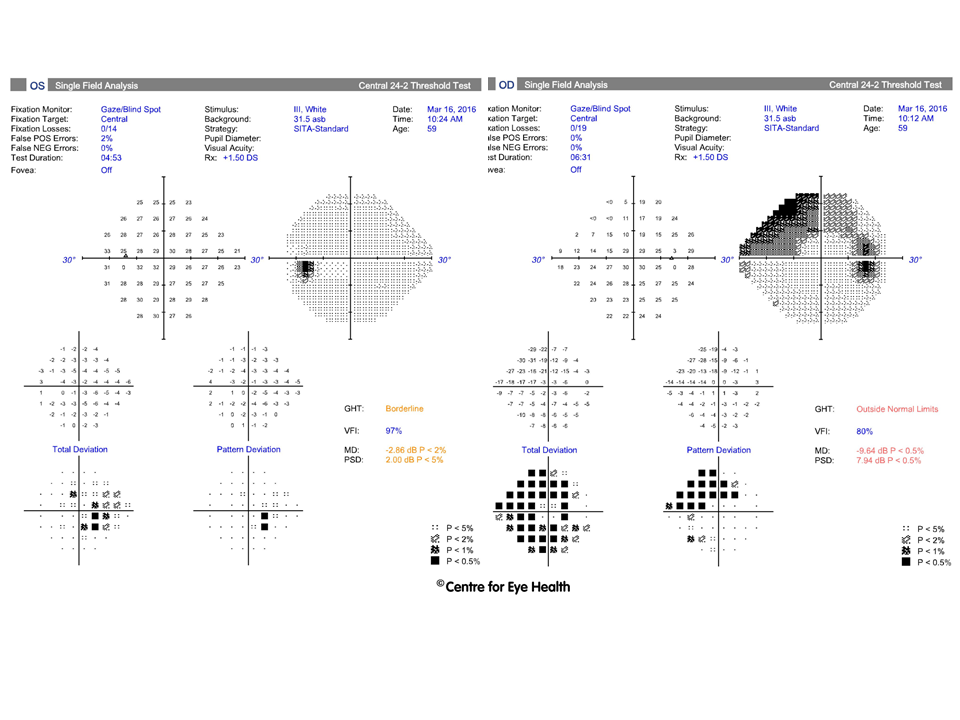)
CFEH Facebook Case #72
Case #72 PDF Download
For the original question click here
Answer
The circular hyper-reflective lesion is actually one of the main retinal vessels traversing the optic cup.
CFEH Facebook Case #71
Case #71 PDF Download
For the original question click here
Answer
Our patient does have glaucoma, probably steroid-related due to prolonged use of nasal steroids. IOP’s are significantly elevated and corneas are thicker than average. Discs are average sized and show large cupping with thin superior and inferior rims. Visual fields show a superior arcuate defect in the left eye. Cirrus RNFL analysis shows thinning of the RNFL in both eyes in a pattern consistent with glaucomatous loss. Nasonex (mometasone) is a steroid based nasal spray used to treat allergies. Recent studies have not shown an association between glaucoma and mometasone. However given there is no family history of glaucoma it is likely that this patient is a steroid responder and the effects of nasal steroids has not been studied on this group.
CFEH Facebook Case #70
Case #70 PDF Download
For the original question click here
Answer
Hereditary haemorrhagic telangiectasia (HHT). The Optomap imaging shows regions of retinal vessel telangiectasia, microaneurysms and ghost vessels in the periphery. Upon further questioning, the patient revealed he had already been previously diagnosed with HHT and was under the care of medical practitioner. HHT is also known as Osler-Weber-Rendu disease and is a rare autosomal dominant condition that results in abnormal blood vessel formation affecting the whole body. These arteriovenous malformations (AVM) occur between arteries and veins, bypassing the capillaries and allowing blood to flow directly into the veins. AVMs are usually fragile and can rupture, resulting in bleeding. This condition can affect the skin, mucous membranes, and organs such as the lungs, liver and the brain. Medical intervention for this condition aims to decrease the degree of haemorrhages and to prevent complications by AVMs. These patients require long term monitoring as lesions can progress, recur or newly manifest as a result of this condition.
CFEH Facebook Case #69
Case #69 PDF Download
For the original question click here
Answer
Congenital grouped pigmentation of the RPE (“Bear tracks”). These are multiple, small, flat, black lesions that are usually clustered in a single quadrant, but in this case are present around 360 degrees of the fundus so this is a somewhat atypical presentation. Usually you would expect the lesions to increase in size towards the periphery of the retina. Fundus autofluroescence shows the lesions to be hypo-autofluorescent which has highlighted them clearly in this case, and OCT imaging shows no obvious abnormalities. This patient had already been screened for bowel disease, however Familial Adenomatous Polyposis (FAP) is typically associated with multiple pisciform lesions such as seen below rather than the bear-tracks seen in this patient.
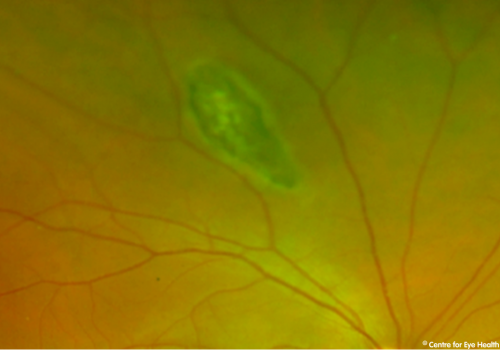)
CFEH Facebook Case #68
Case #68 PDF Download
For the original question click here
Answer
Astrocytic hamartoma. This patient has a raised white-yellow lesion with a pigmented border, located supero-temporally, adjacent to a choroidal naevus 2-3 disc diameters in size. OCT scans indicate the raised lesion is intra-retinal with shadowing on OCT. These findings suggest a diagnosis of astrocytic hamartoma. An astrocytic hamartoma is a benign tumour originating from the retinal nerve fibre layer which can involve most layers of the retina. These growths specifically involve the enlargement and proliferation of astrocytes, which are supportive glial cells. They can present as an elevated, opaque, white nodule or as a flat and semi-translucent lesion with undefined margins. These lesions may be calcified or non-calcified. It is thought that the nodular appearance of the lesion eventually progresses to a mulberry appearance, which indicates increased calcification. Isolated, unilateral astrocytic hamartomas found in healthy individuals usually occur spontaneously. These lesions do not commonly progress or result in any complications. They are usually not associated with any other ocular findings and do not require any treatment. Multiple, bilateral or larger-sized retinal astrocytic hamartomas may be systemically associated with neurocutaneous syndromes such as tuberous sclerosis and neurofibromatosis. Rarely, astrocytic hamartomas can grow aggressively. This can result in exudative retinal detachments. In some cases, surgical interventions, radiation therapy or enucleation may be necessary. Despite the isolated single nature of the astrocytic hamartoma, given that this is a rare condition and there are potential systemic associations, referral to a retinal specialist for confirmation of diagnosis and management is recommended.
CFEH Facebook Case #67
Case #67 PDF Download
For the original question click here
Answer
These findings indicate an acquired vitelliform lesion in the right eye. There are also hypo-autofluorescent patches at the posterior pole. The aetiology of these changes is unclear however they may indicate previous central serous chorioretinopathy. There is no sign of CNV in this eye. Analysing the imaging that lead us to this conclusion, we can see that there is a well-defined round yellow lesion at the macula (the vitelliform lesion). This corresponds to the hyper-reflective mass seen on OCT in the RPE with overlying pigment migration. Fundus autofluorescence (FAF) showed distinct hyperfluorescence of this lesion as well as subtle areas of hypofluorescence at the posterior pole. In earlier stages, the vitelliform material appears as an area of round hyper-autofluorescence on FAF. Over time, FAF is visible mainly in the inferior portion of the lesion. Eventually, the lesion reaches the vitelliruptive scrambled egg stage and hyper-autoflourescence is concentrated at the borders of the lesion. In the final, atrophic stage there is a large reduction in FAF and the lesion appears hypo-autofluorescent.
CFEH Facebook Case #66
Case #66 PDF Download
For the original question click here
Answer
The patient has Salzmann’s nodular degeneration. Also present are senilius, and (not visible in these photos) bilateral Hudson-stahli lines and epithelial basement membrane dystrophy. Salzman’s nodular degeneration is a rare degenerative condition that is slowly progressive. It is typically bilateral (63% of cases) and can be found in a wide range of ages (cases have been reported from the age of 4 through to 83 with an average age of diagnosis of 59 years). Studies have shown a high association between Salzmann’s nodular degeneration and both osteoporosis and ocular surface disease or corneal “insult”. Symptoms experienced are usually due to the raised nature of the lesions and can include dry eye, foreign body sensations and recurrent corneal erosions. Arcus senilius is a deposit of lipid in the peripheral corneal stroma and is not an uncommon finding in this age group. Epithelial basement membrane dystrophy (EBMD) was reviewed a few weeks ago in case 52 (please refer to that case for further information).
CFEH Facebook Case #65
Case #65 PDF Download
For the original question click here
Answer
An artefact induced by a posterior chamber IOL implanted following recent monocular cataract surgery.
CFEH Facebook Case #64
For the original question click here
Answer
The multiple large, raised lesions in the right eye are consistent with sclerochoroidal calcification. There is also non-specific pigmentary changes at the posterior pole. Sclerochoroidal calcification is characterised by benign yellow-white sub-retinal lesions, typically found along the vascular arcades. They are most commonly found in elderly Caucasians. Extended depth OCT imaging has shown these lesions originate from the sclera with thinning of the overlying choroid, as seen in this patient. Lesions have a typically irregular or “rocky” contour, unlike a choroidal naevus or small melanoma which will typically have a gentler, more uniform slope. Fundus autofluorescence showed mixed hyper- and hypo- autofluorescence of the pigmentary changes noted at the posterior poles. The elevated lesions were hyperfluorescent which is a feature typical of sclerochoroidal calcification. Usually fundus hyper-autotfluoresence is associated with increased lipofuscin, however in this condition it has been proposed that the thinning choroid allows the underlying sclera to hyper-autofluoresce. While most presentations of this condition are idiopathic, some have systemic associations. A patient diagnosed with sclerochoroidal calcification should be referred for a systemic work-up to screen for calcium-phosphate metabolic abnormalities or renal tubular hypokalemic metabolic alkalosis. There have also been some case reports of parathyroid adenoma with calcium elevation being detected following a diagnosis of sclerochoroidal calcification.
CFEH Facebook Case #63
Case #63 PDF Download
For the original question click here
Answer
Isolated choroidal melanocytosis. This condition falls within the spectrum of oculo (dermal) melanocytosis but affects only the choroid, not the uvea and episclera. Oculodermal melanocytosis typically does affect these other areas as well and is characterised by pigment elsewhere in the uveal tract, or cutaneously in the periocular area (nevus of Ota – imaged below):
While isoloated choroidal melanocytosis is rare, a series of 11 cases has been reported in the literature by Ausberger et al. The authors described the clinical characteristics of this condition as “an homogenous area of dark brown pigmentation greater than 5mm in diameter, with complete lesion flatness”, differentiating it from choroidal naevi (typically less than 5mm diameter) and melanocytomas (which are raised). It is estimated that one in 400 Caucasian patients with ocular melanocytosis will develop uveal melanoma. Unfortunately however, the increased fundus pigmentation in patients with choroidal melanocytosis can make it more difficult to detect a small pigmented melanoma. Augsberger et al. proposed that, based on their histological analysis of the lesions, the increased risk of melanoma in these cases is proportional to the percentage of uvea involved. This would mean that the risk of developing choroidal melanoma in cases such as this is only slightly higher than the general population. Patients with choroidal melanocytosis require annual monitoring to ensure the early detection of melanotic changes.
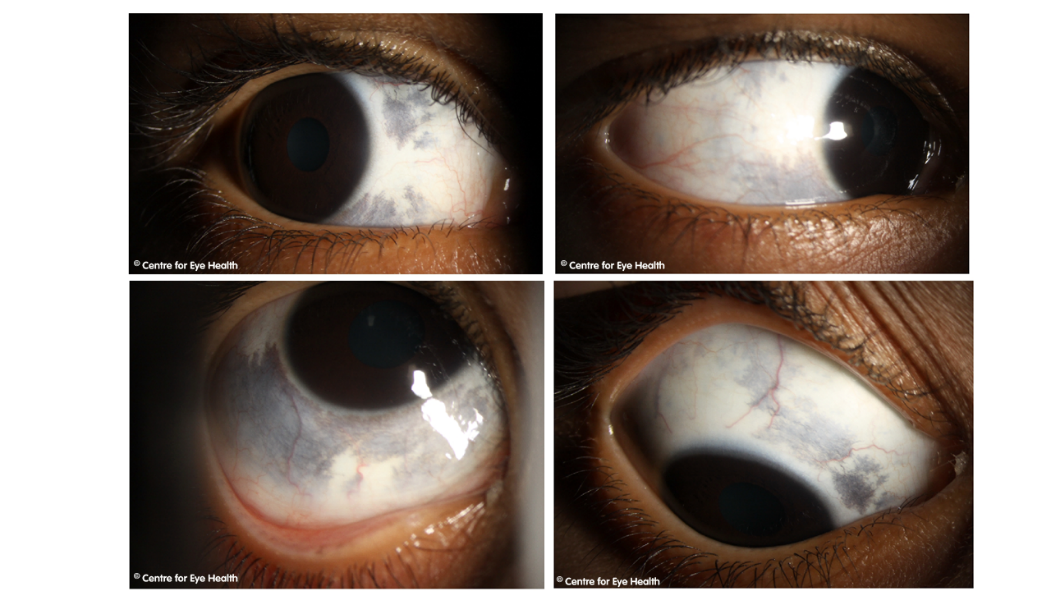)
CFEH Facebook Case #62
Case #62 PDF Download
For the original question click here
Answer
There are several pathological processes going on in this patient: 1. The marked tortuosity and dilated nature of the veins may indicate a pending central retinal vein occlusion 2. Pachychoroid spectrum disease. The large diameter of the hyper-autofluorescence indicates that this may be a chronic CSCR and the small serous PED surrounding subretinal fluid, indicates active disease. Measuring the patient’s blood pressure in-office found it to be 160/92. The retinal findings and elevated blood pressure measurements should be communicated to the patient’s GP in a timely fashion with concurrent referral to an ophthalmologist. For more information on hypertensive retinopathy, possible sequelae and management, please refer to CFEH’s new chairside reference, released June 2017 which covers all you need to know, go to: https://centreforeyehealth.com.au/wp-content/uploads/2017/06/Chairside-reference-Hypertension.pdf For further information on pachychoroid spectrum disease, refer to facebook case 55.
CFEH Facebook Case #61
Case #61 PDF Download
For the original question click here
Answer
Reticular macular disease (reticular pseudodrusen) Colour fundus photography and red free imaging showed a reticular pattern of small, yellow-white, round and/or oval lesions known as reticular pseudodrusen (RPD). Using Infrared imaging (adjacent to OCT line scans), RPD appear as groups of hypo-reflective lesions with larger lesions accompanied by a halo-like appearance or “target” aspect. On OCT, RPD appear as well-defined, hyper-reflective deposits beneath the RPE and ELM or OPL. RPD is frequently found in eyes with AMD but less frequently than soft drusen. The risk factors associated with RPD are similar to those of AMD and include age, gender, smoking and genetic risk factors. Recent studies have shown that the presence of RPD was a strong risk factor for progression to late-stage AMD and that showed that RPD are associated more strongly with geographic atrophy than with choroidal neovascularisation. In this patient, other early structural changes that may precede the development of late AMD were also identified including hyper-reflective foci, subsidence of the outer plexiform layer/inner nuclear layer, hypo-reflective wedge-shaped bands consistent with nascent geographic atrophy, sub-RPE hyper-reflective columns and disruption in the ellipsoid zone and external limiting membrane overlying the drusen.
CFEH Facebook Case #60
Case #60 PDF Download
For the original question click here
Answer
OCT imaging of the macula shows increased reflectivity on the posterior cortical vitreous extending from the optic nerve head to the macula. This increased reflectivity is causing the ERM-like appearance seen in the macular photo. There are also some glial remnants visible at the optic nerve head (Bergmeister’s papillae – please refer to Facebook case 28 for further information on this condition). These findings indicate the presence of persistent hyaloid remnants on the posterior cortical vitreous of the right eye. Persistent remnants of the hyaloid vascular system has been reported in 3% of infants born full-term, and 95% of those born premature. These remnants can often be seen as; Mittenforf dots on the posterior lens capsule (remnants of the anterior portion of the hyaloid artery), a persistent pupillary membrane or as Bergmeisters papille at the optic disc. The presentation here is more unusual, but not problematic. Routine review is all that is required.
CFEH Facebook Case #59
Case #59 PDF Download
For the original question click here
Answer
The anterior eye photos show left iris hypoplasia and polycoria as well as an inferotemporal corneal opacity. Gonioscopy shows some superotemporal pigment deposition on the endothelium and temporal and inferior synechiae. The trabecular meshwork was visible in at least two quadrants. Possible causes include trauma and ICE syndrome, although the patient denies any history of ocular trauma or surgery. ICE syndrome is typically unilateral and is characterised by corneal oedema and iris abnormalities, including atrophy, holes, nodules and pupillary distortion. It is caused by the proliferation of abnormal corneal endothelial cells. These cells grow to form a membrane over the iris and anterior chamber angle. Over time, the contraction of this membrane causes the iris abnormalities and secondary angle closure glaucoma results. Management of secondary glaucoma associated with this condition is difficult as it is often complicated by corneal oedema.
CFEH Facebook Case #58
Case #58 PDF Download
For the original question click here
Answer
This patient has had a branch retinal vein occlusion some time ago, and subsequently has developed neovascularisation and associated fibrosis which is showing up on OCT as a raised, hyper-reflective lesion above the plane of the retina. Hypertension and hyperlipidemia can cause areteriolar sclerosis, resulting in compression of the vein at arteriovenous crossings. Other conditions associated with BRVO include diabetes mellitus and peripheral arterial disease. Occlusion of one of the major branch retinal veins (major BRVO) may be either ischemic (2/3 of cases) or non-ischaemic (1/3 of cases) and 65% of BRVO’s are found in the superior-temporal quadrant. Neovascularisation can develop following an ischaemic BRVO, such as in this case. The potential complications of a BRVO include macular oedema, retinal neovasuclarisation, vitreous haemorrhage and tractional retinal detachment, so treatment is aimed at managing these complications to improve visual acuity and reduce metamorhopsia.
CFEH Facebook Case #57
Case #57 PDF Download
For the original question click here
Answer
The findings are most consistent with congenital stromal corneal dystrophy, which has previously been referred to as “congenital hereditary stromal dystrophy” This is an autosomal dominant dystrophy that is either non-progressive or slowly progressive. Typically, diffuse bilateral corneal clouding is noted with flake-like stromal opacities as seen in the corneal photos. The corneal stroma is affected throughout and anterior corneal surface is unaffected. As is typical, in this case the anterior OCT shows an increased stromal reflectivity in both eyes. Usually, pachymetry will show a thickened cornea, however in this case the cornea is actually at the thin end of normal. Examination of immediate family members (mother and 2 brothers) revealed a similar corneal appearance in each:
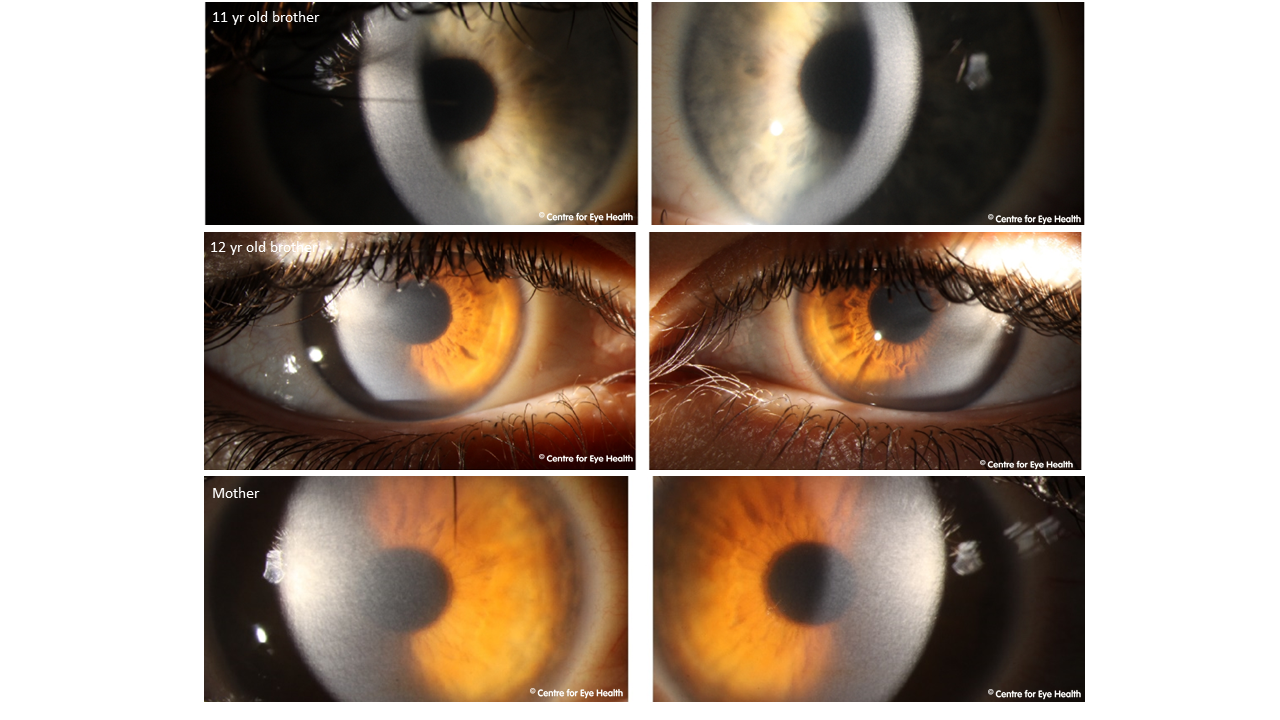)
CFEH Facebook Case #56
Case #56 PDF Download
For the original question click here
Answer
Bull’s eye maculopathy. Depigmentation surrounding the fovea can be seen in the retinal photos. OCT imaging shows a marked thinning of the ONL, ELM, ISe and RPE parafoveally. Autofluorescence imaging showed annular hypo-autofluorescence surrounding the fovea with adjacent hyper-autofluorescence both within and outside the hypofluorescent annulus. A bull’s eye maculopathy is commonly associated with retinal toxicity (chloroquine, hydroxycholoquine, clofazimine) however in this case the patient denied previous use of medications known to cause toxicity. Given this, and the longstanding nature of the night vision problems, the most likely cause is a retinal dystrophy – cone dystrophy, cone-rod dystrophy or Stargardt disease, although the latter is usually associated with retinal flecks so unlikely here. Referral to a retinal specialist was recommended.
CFEH Facebook Case #55
Case #55 PDF Download
For the original question click here
Answer
The notable findings on funduscopy (and seen in the retinal image) include areas of mottled RPE change within and around the macula and near the optic nerve. Fundus Autofluorescence (FAF) shows mixed stippled hyper- and hypo-autofluorescence corresponding to these areas of RPE change. Note that these changes are more evident using fundus autofluorescence than with funduscopy/photography. OCT imaging showed an isolated shallow PED and disruption of the ISe zone with some overlying pigment migration at this point (seen as a hyper-reflective foci on the OCT scan). These findings are consistent with pachychoroid pigment epitheliopathy (PPE), which is part of the Pachychoroid spectrum of disease. This spectrum is characterised by the common features of increased choroidal thickness, reduced fundus tessellation, drusenoid RPE changes, areas of hyper and hypo autofluorescence that are in excess of RPE changes noted clinically, and the presence of small PEDs overlying areas of thickened choroid. In addition to PPE, other conditions included in the spectrum include central serous chorioretinopathy (CSCR), polypoidal choroidal vasculopathy (PCV) and pachychoroid neovasculopathy. PPE is likely to be a “forme fruste” manifestation of CSCR as it involves no history of serous macular detachment or sub-retinal fluid but other clinical characteristics are similar. A recent study showed that in eyes with pachychoroid spectrum disease, the finding of shallow irregular PEDs on OCT (such as in this case), may be a significant indicator of the development of type 1 neovascularisation.
CFEH Facebook Case #54
Case #54 PDF Download
For the original question click here
Answer
There is an anomalous blood vessel which appears to loop and corkscrew around on the nasal aspect of the right disc, suggestive of a shunt vessel. Spectralis OCT through the optic nerve shows a focal hyper-reflective lesion which appeared co-localised with the vascular anomaly seen on the retinal images. An OCT through the right macula showed a number of cystic spaces, consistent with cystoid macular oedema, and accounting for the reduced vision. It also shows a hyper-reflective lesion adjacent to the cystic space at the macula. This is another anomalous blood vessel, visible during the examination but not clear on the Optomap image. Optomap imaging shows numerous microaneurysms and dot/blot haemorrhages almost 360 degrees in the peripheral retina, a finding present in both eyes. Incidentally, peripheral examination also showed a raised area superiorly to superotemporally in the right eye. Spectralis OCT through this region (below) confirmed the presence of a retinoschisis.
This patient was referred to a retinal specialist ophthalmologist for and opinion and a review of the patient’s vascular risk factors with his GP was also recommended. The anomalous shunt vessel is hypothesized to have developed secondary to a retinal vein occlusion.
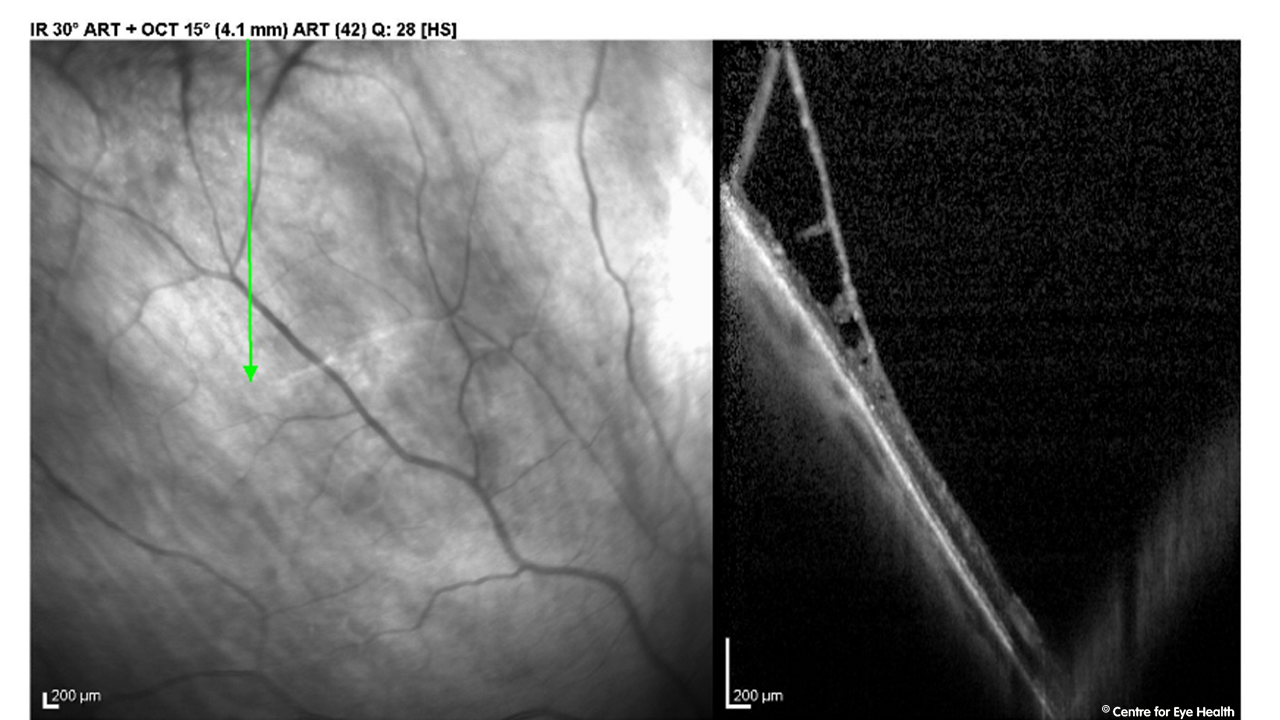)
CFEH Facebook Case #53
Case #53 PDF Download
For the original question click here
Answer
Posterior polar annular choroidal dystrophy (PPACD) PPACD is a rare condition characterised by progressive chorioretinal atrophy around the vascular arcades and optic disc. There is typically some preservation of the choriocapillaris at the junction between the area of atrophy and normal which appears as a fringe of hyper-autofluorescence. The OCT images show attenuation of the outer retinal layers with thinning of the outer nuclear layer and an absent IsE zone. Electrophysiology testing showed findings consistent with PPACD. A full field ERG showed the photopic responses (cone responses) to have slightly delayed b wave of reduced amplitude in both eyes. Pure rod responses were substantially reduced in amplitude with prolonged a and b waves (see image below).
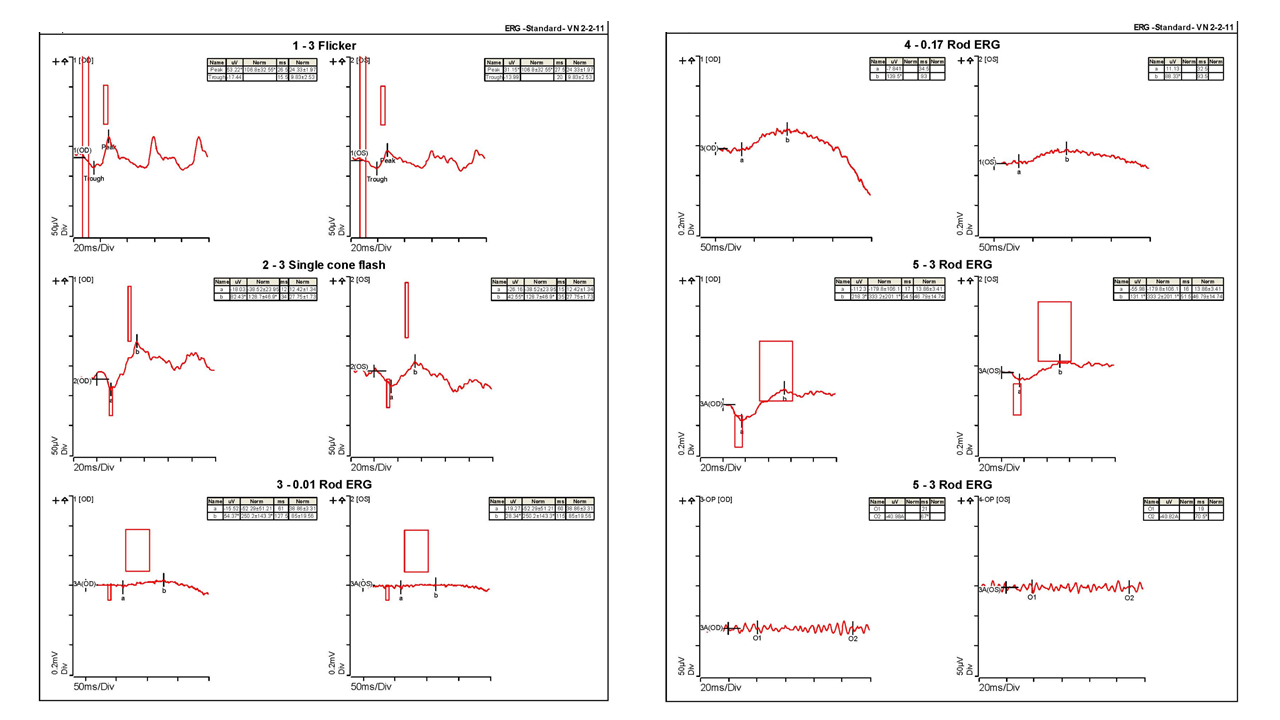)
CFEH Facebook Case #52
Case #52 PDF Download
For the original question click here
Answer
Epithelial basement membrane dystrophy (EBMD) – formerly known as map-dot fingerprint dystrophy or Cogan microcystic epithelial dystrophy. This condition is the most common type of corneal dystrophy, affecting approximately 2% of the population. EBMD has a spectrum of possible presentations, including this one where the patient has “dots”, characterised by round or oval intraepithelial opacities that have a greyish appearance and don’t stain with fluorescein dye. The anterior OCT shows corresponding abnormalities at the basement membrane. Other presentations can include “maps”, “fingerprint” lines or a “bleb” pattern. Onset is typically in adult life and the condition is often asymptomatic initially but can be associated with painful corneal erosions and visual disturbances (irregular astigmatism and image ghosting). This condition may be degenerative. Initially management of the corneal erosions may include bandage soft contact lenses, lubricating gels or hyperosmotic ointment. More advanced cases may require greater intervention, such as superficial keratectomy.
CFEH Facebook Case #51
Case #51 PDF Download
For the original question click here
Answer
The patient was referred urgently to the local public hospital ophthalmology clinic where he was diagnosed with posterior scleritis and treated with steroids and NSAIDs. The scleritis was thought to possibly be related to a systemic connective tissue disorder. Scleritis may be due to infection, autoimmunity or trauma with an autoimmune aetiology being most common. Scleritis is associated with underlying systemic disease in approximately 36–57% of patients. The underlying systemic disease is most often rheumatoid arthritis. Scleritis is a painful, destructive inflammation deep in the sclera that produces uveal effusion and secondary angle-closure glaucoma. It can potentially cause permanent damage to the eye and vision, however early treatment has been shown to be effective in controlling inflammation and limiting visual loss. It can occur in all age groups, with a median age of 50 years. Posterior scleritis may present with a wide range of clinical signs and symptoms that include periocular pain, headache, pain on eye movement and reduced vision. Clinical findings associated with this condition can include fluid in the Tenon capsule (episcleral space), swelling of the optic disc, a distended optic nerve sheath, retinal detachment and scleral nodules however 17% of cases show no abnormal ocular signs(nodular scleritis). Up to 60% of patients have associated anterior scleritis identified at some stage of the disease process. High penetration OCT and B-mode ultrasonography have been used to show a thickening of the choroid during acute posterior scleritis, decreasing over time with treatment and thickening again with reactivation of the disease.
CFEH Facebook Case #50
Case #50 PDF Download
For the original question click here
Answer
Peripheral retinal examination revealed proliferative diabetic retinopathy (PDR) with a large neovascular sea-fan in the superotemporal arcades not apparent in the posterior pole images. Extensive peripheral haemorrhages, venous calibre abnormalities and retinal ischaemia were also present. The posterior pole image and OCT line scans showed moderate diabetic macular oedema (International Clinical Disease Severity Scale for Diabetic Retinopathy and Diabetic Macular Edema). By examining the posterior pole in isolation, the level of retinopathy would likely have been significantly underdiagnosed as moderate NPDR with macular oedema. This case highlights the need to examine the peripheral retina in all patients with diabetes.
A recent study in the UK (Talks et al. 2015) has shown that 24% of neovascularisation in diabetic patients occurs outside the area of the posterior pole imaged by two 45 degree images centred around the optic nerve and macula. This indicates that a quarter of all neovascularisation in diabetic patients may be missed by relying on photography of the posterior pole alone. Further studies have indicated that individuals with peripheral diabetic lesions have a greater risk of DR progression. For a summary of the latest literature on this topic, you here.
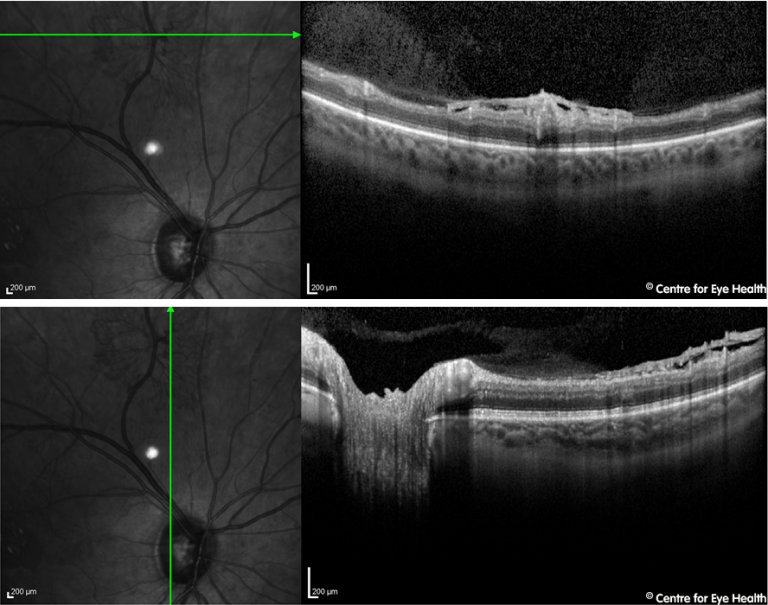)
CFEH Facebook Case #49
Case #49 PDF Download
For the original question click here
Answer
A retained foreign body – the initial injury was caused by a hammer that chipped and the foreign body was never removed. Imaging shows a markedly elevated lesion inferior to the left optic disc with associated retinoschisis, vitreous traction and inferior retinal atrophy. The B-scan ultrasound image below is consistent with a retained foreign body showing posterior shadowing inferior to the disc:
This patient also had narrow, occludable angles and was referred to a retinal specialist.
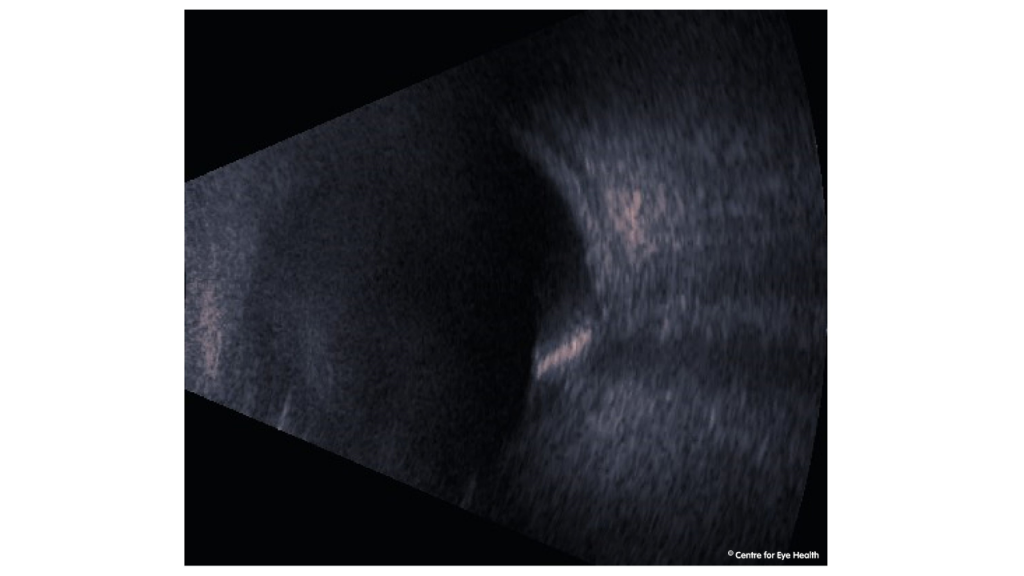)
CFEH Facebook Case #48
Case #48 PDF Download
For the original question click here
Answer
Dominant optic atrophy (DOA), also known as dominantly inherited juvenile optic atropy (DIJOA) or autosomal inherited optic atrophy. Both discs show temporal pallor with shallow “saucerised” cupping of the inferior, superior and temporal neuro retinal rims. These findings are characteristic of optic atrophy. There is diffuse thinning of the RNFL as seen on red-free, more notable in the left eye. The supero- and infero- temporal quadrants show marked thinning compared with the normative database on Cirrus, and ganglion cell analysis shows marked thinning that is symmetrical between the two eyes. DOA is an inherited bilateral optic neuropathy. Presentation is usually in the first or second decade of life, however it can present later in life (as in this case). Presenting signs and symptoms include a slow, progressive, insidious decrease of vision. The degree of loss can vary significantly both between and within families, with vision usually in the range of 6/9 – 6/30. Inheritance is autosomal dominant with penetrance of 43-100%, depending on the family. The main gene responsible is OPA1, this being the affected gene in 75% of cases. 20% of cases of DOA are “syndromic”, meaning they have associated systemic signs, including neurosensory deafness, myopathy, peripheral neuropathy, stroke, multiple sclerosis or progressive external ophthalmoplegia.
CFEH Facebook Case #47
Case #47 PDF Download
For the original question click here
Answer
This patient has notable thinning of both the superior and inferior neuro retinal rim and retinal nerve fibre layers in the right eye. A tentative diagnosis of normal tension glaucoma was proposed, however given the low risk profile in conjunction with the IOP and other clinical results being symmetrical, the patient was referred for an MRI to rule out neurological causes. The MRI results showed a right pituitary adenoma with associated cystic degeneration. The patient was subsequently referred to a neurologist and an endorcrinologist. The clinical pearl here is that in cases of asymmetrical RNFL loss, neurological causes must be excluded.
CFEH Facebook Case #46
Case #46 PDF Download
For the original question click here
Answer
Bietti Crystalline Dystrophy. Bietti crystalline dystrophy is characterised by the presence of numerous glistening yellow crystalline deposits distributed throughout the fundus and occasionally also present in the superficial cornea near the limbus. OCT investigations show these deposits may be located throughout all retinal layers but are most commonly found adjacent to the inner side of the RPE. The deposits are less apparent in areas of pigment epithelial atrophy. A characteristic feature of Bietti crystalline dystrophy is the presence of circular hypo-reflective areas within the outer nuclear layer, termed tubular degeneration or tubular formation. These areas are typically found where there is damage to the RPE and rarely seen where the RPE is intact. In this case, the deposits are predominantly found in the outer retinal layers. There is loss, attenuation and disruption of the ISe zone, outer retinal layers and choriocapillaris and tubular degeneration can be identified (circled in red on OCT image below).
Fundus autofluorescence highlights the multiple areas of geographic atrophy in this patient. These areas appear densely hypo-autofluorescent with speckled hypo-autofluorescence surrounding them as well as radiating branches of hyper-autofluorescence. Bietti crystalline dystrophy usually shows an autosomal recessive inheritance pattern and is most common in East Asian populations, particularly those from China and Japan. Presentation is usually in the 4th or 5th decade of life when symptoms start to develop. Presenting symptoms are typically a slow progressive vision loss and nyctalopia. As the condition progresses there is a diffuse atrophy of the choriocapillaris and decrease in size and number of the crystals. The atrophic areas progressively become confluent and extend into the peripheral retina with the end result being diffuse chorioretinal atrophy. The rate of progression and severity of this condition is quite variable. This patient was referred to a retinal specialist.
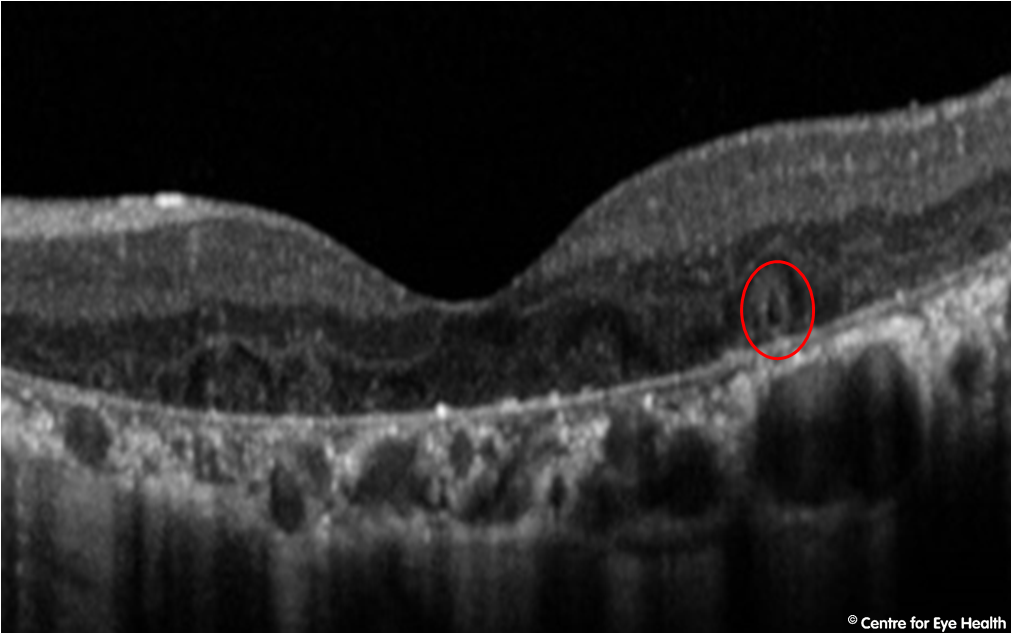)
CFEH Facebook Case #45
Case #45 PDF Download
For the original question click here
Answer
Maternally Inherited Diabetes and Deafness (MIDD) This condition is caused by a point mutation in the mitochondrial DNA and is thought to be responsible for between 0.5 and 2.8% of diabetes. It is characterised by maternally inherited diabetes (85% penetrance) and impaired hearing, usually in the high frequencies. The hearing loss is sensorineural and originating from the cochlear rather than the cranial nerve. MIDDS is often accompanied by a pattern macular dystrophy. The macular dystrophy is characterised by both pigmented retinal lesions and atrophy of the RPE and/or choroid. The pigmented lesions range from appearing small and found at the macula through to radiating patterns surrounding the macula or optic nerve. Visual symptoms can include vision loss, photophobia, night vision problems and scotomas. The OCT images from this patient show some mottling of the RPE and a left micro-hole at the macula with some mottled hyper-autofluorescence, in particular at the fovea. The age of onset of MIDD is typically between 25 and 35 years of age however it can range from 11 years to 68 years. Onset is usually insidious (such as in Type 2 diabetes), however about 20% of cases have an acute presentation with ketoacidosis occurring in around 8% of cases. The mitochondrial mutation causing this condition is also responsible for other systemic manifestations, including MEALS (mitochondrial encephalomyopathy, lactic acid and stroke-like episodes); psychiatric disorders (depression, schizophrenia and phobias); myopathy of the proximal limb muscles; endocrine disease, renal disease and rapid onset heart failure in young adults. MIDD increases the risk of developing these conditions. It was recommended this patient undergo electrophysiological testing to confirm a diagnosis of macular dystrophy. She was also referred to a retinal specialist for confirmation of a diagnosis.
CFEH Facebook Case #44
Case #44 PDF Download
For the original question click here
Answer
Axenfeld-Reiger Syndrome. This syndrome is primarily an eye condition but may have systemic manifestations. It affects 1 in 200 000 people and has an autosomal dominant inheritance pattern. Ocular manifestations can include iris atrophy, corectopia (displacement of the pupil), polycoria, corneal abnormalities and hypertelorism (an abnormally increased distance between the eyes). This patient has a large corneal diameter, pupil miosis, superior-nasal displacement of both pupils (more prominent before dilation), left pseudopolycoria, posterior embryotoxin and a moth-eaten appearance to the irides. This anterior segment dysgenesis is consistent with Axenfeld-Reiger syndrome. The synechiae noted on gonioscopy are likely to have resulted from prior surgeries. Optic nerve head evaluation showed thinning of the neuro-retinal rim and enlargement of the cup, while Cirrus OCT shows a marked thinning of the RNFL that is characteristic of glaucoma. Additional systemic manifestations can include:
- A flattened midface with broad flat nasal bridge and prominent forehead
- Microdontia (one or more teeth are smaller than normal)
- Oligodontia (congenital absence of some teeth)
- Redundant periumbilical skin (extra folds of skin around the belly button)
- Heart defect
- Hypospadias
- Anal stenosis
- Pituitary gland abnormalities which can slow growth
CFEH Facebook Case #43
Case #43 PDF Download
For the original question click here
Answer
Differentials based on the information given include small crowded discs, buried disc drusen, optic neuritis, optic atrophy and anterior ischaemic optic neuropathy. Further imaging showed the following:
In this patient, the OCT line scans show an elevation of both optic nerves, however the Cirrus OCT also shows a thinning of the retinal nerve fibre layer in most quadrants as opposed to a thickening as you see in true papilloedema. The fundus autofluoresence shows hyper-reflective “lumps” on the optic nerve head (easier to see on the left eye which has been included here for illustrative purposes). This is a finding characteristic of optic nerve head drusen. Optic nerve drusen occur in approximately 1-2% of the general population, and are usually bilateral (86% of cases). The drusen are typically calcified deposits within the optic nerve head and may be buried or obscured beneath the disc surface (such as in this case), causing pseudopapilloedema. Drusen may enlarge slowly over time and visual field defects are often progressive but they rarely affect functional vision. This patient however was referred to a neuro-ophthalmologist, due to the extent of the field defect at this time. Although no definitive treatment has been identified, IOP reduction may sometimes be used to try and slow the progression of field loss.
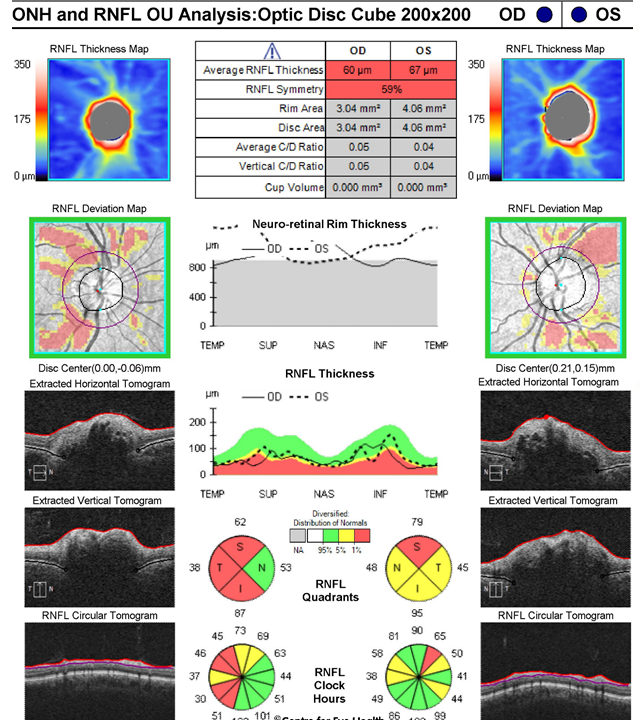)
CFEH Facebook Case #42
Case #42 PDF Download
For the original question click here
Answer
The patient had conductive keratoplasty (CK), a procedure introduced in 2002 which aims to correct low hyperopia (ideally around +1.50D), or reduce the dependence on reading glasses for presbyopes. In this procedure, the cornea is treated with low level radio-frequency energy in specific areas of the peripheral cornea. The areas treated contract in response, causing a steepening of the cornea thereby reducing hyperopia and/or creating a small amount of myopia to assist with reading. There is typically some regression with time, the procedure often may need to be repeated and is not currently widely used. The reduced vision, however is caused by likely choroidal neovascularisation at the macula. While the retinal photo does not show marked abnormalities, the OCT line scan show the presence of sub-retinal and intra-retinal fluid, general retinal disorganisation as well as fibrosis, indicative of CNV. This case illustrates the value of OCT imaging in assisting the timely detection of eye disease.
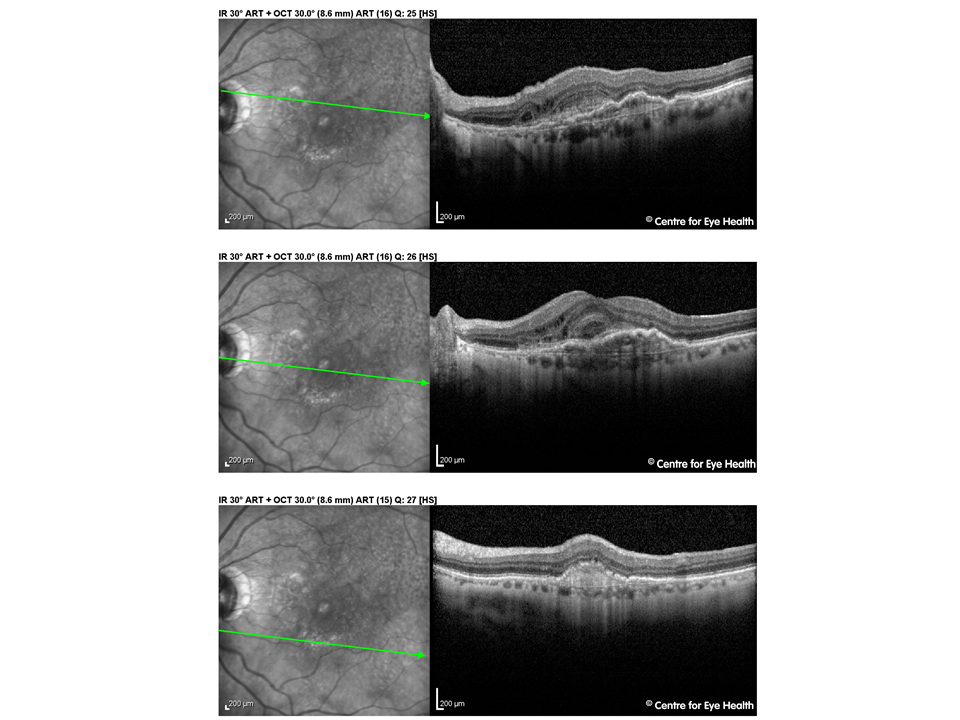)
CFEH Facebook Case #41
Case #41 PDF Download
For the original question click here
Answer
Macular Telangiectasia type 2 (Mac Tel 2), stage 2. The OCT image in the right eye shows disruption of the outer retinal layers with some cystic spaces at the nasal aspect of the lesion. The RPE/Bruchs membrane complex is very irregular in the affected region and the pigment plaque in this eye corresponds with increased intraretinal reflectivity. Looking at the left macular OCT, there is also some disruption of the outer retinal layers, including disruption of the ellipsoid zone. Mac Tel 2 is a bilateral perifoveal vasculopathy of unknown cause, however a genetic component to this disease is highly likely. Prevalence is 0.1% of the general population. Symptoms typically start in the 5th or 6th decade of life and the early signs may be very subtle. Signs and symptoms are typically limited to mildly reduced visual acuity and metamorphopsia, both of which are subtle at first but increase as the disease progresses. Typical clinical findings include a dulling or loss of the foveal reflex, a greying of the parafoveolar area, small foveal cystoid changes and pigment clumping. A pseudo-vitelliform lesion can also form in the central macula. Fundus autofluorescence imaging can also be useful in evaluating MacTel 2. Areas of photoreceptor atrophy appear hyperfluorescent when a pseudovitelliform lesion develops and RPE atrophy appears hypofluorescent. The normal central attenuation of FAF may be lost in eyes with MacTel2 .
The literature identifies 5 stages of the disease process with stage 5 (proliferative stage) characterised by the formation of subretinal neovascularisation. This stage is accompanied by retinal oedema, yellow exudates, subretinal / intraretinal haemorrhages and, over time, retinal fibrosis. There is no treatment for the pre-proliferative stages, however photodynamic therapy may be useful in decreasing vision loss in the proliferative stages.
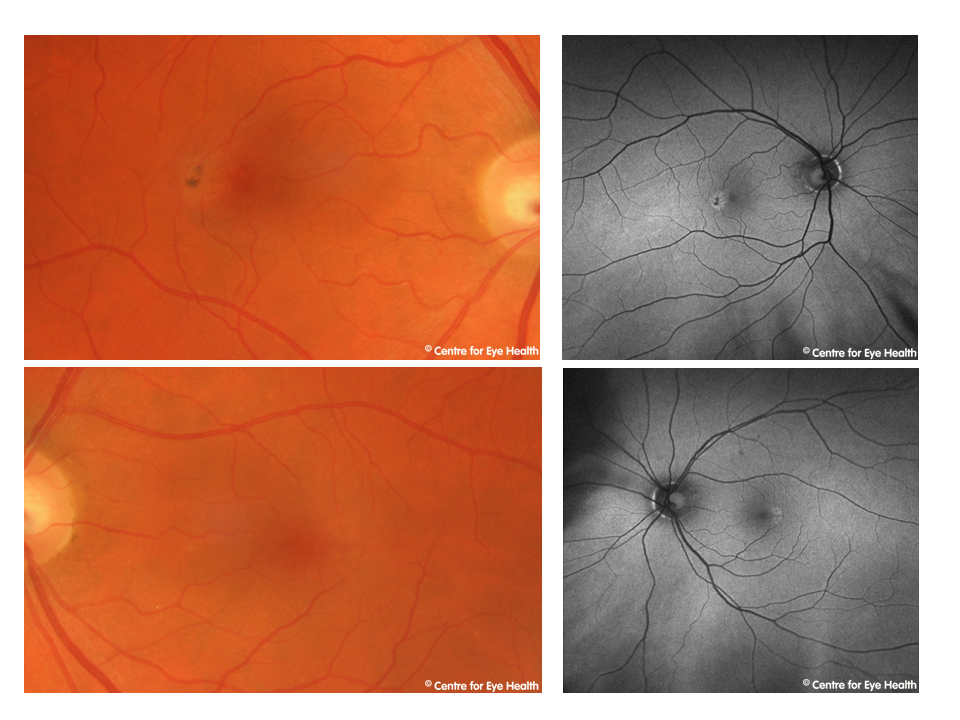)
CFEH Facebook Case #40
Case #40 PDF Download
For the original question click here
Answer
The appearance of the lesion is consistent with solitary idiopathic choroiditis (SIC), however further assessment by a retinal specialist was recommended to confirm this. Solitary idiopathic choroiditis may appear at any age, however patients are typically Caucasian and present between 20 and 50 years of age. Visual acuity is usually normal, unless the lesion affects the foveal region. Differential diagnoses include amelanotic choroidal tumour (such as melanoma, naevus and osteoma) and ocular inflammation such as sarcoidosis, tuberculosis and toxocariasis. The appearance of the SIC lesion may vary, depending whether it is active or inactive. Inactive lesions typically appear as a discrete, round yellow-white lesion that has a characteristic orange-yellow halo around the lesion, as can be seen in this patient. Fundus autofluorescence imaging typically shows homogenous hyper-autofluorescence and OCT imaging shows these lesions to be smooth and dome-shaped with the overlying choroid appearing compressed or thinned. Recent studies using enhanced depth imaging OCT now suggest that the lesions may extend into the sclera and are not just limited to the choroid as previously thought. Active lesions appear a dull yellow colour with ill-defined margins, sub-retinal fluid with yellow intra-retinal exudative material. Focal haemorrhages may occasionally also be noted. This case was referred to ophthalmology for confirmation of the diagnosis.
CFEH Facebook Case #39
Case #39 PDF Download
For the original question click here
Answer
This patient has a nasal staphyloma as seen in the OCT image. With her prescription which corrects for her foveal vision in place – as typically recommended by the instrument’s algorithm – the light falling on the nasal retina would be defocussed as the retinal plane is bowed posteriorly in the region of the staphyloma (by definition). This has caused a corresponding apparent temporal field depression in the initial visual field examination. Upon re-examination, an additional -2.00DS was added to the patient’s prescription for the purposes of the field test. This focussed the light more accurately in the area of the staphyloma, as the light rays are more diverged, causing the apparent temporal depression to disappear. However the light falling on the temporal (non-staphylomatous) retina is now defocussed, with light rays too divergent for the retinal plane in this region, causing an apparent nasal field depression instead. Thus, a simple increase in myopic correction to correct for different retinal planes has caused an apparent reversal of the field defect. One differential for a temporal field defect such as was seen on initial examination would be a chiasmal lesion. Demonstration of reversal of a field defect such as in this case, would suggest that that defect is likely to be optical in origin rather than pathological.
CFEH Facebook Case #38
Case #38 PDF Download
For the original question click here
Answer
Solar retinopathy. Solar retinopathy is a photochemical retinal injury caused by either direct or indirect viewing of the sun. The risk of damage is higher in patients taking drugs causing photosensitivity such as tetracycline, isoretinoinand psoralens, while it is reduced in patients with darkly pigmented retinae and those with high refractive errors. In this case the patient in question was suffering from a mental disturbance that caused him to stare at the sun for 3 hours 20 minutes approximately 3-4 months prior to his examination at the Centre. Symptoms of solar retinopathy include decreased vision (in the range 6/7.5 – 6/30) with associated central scotomas. Metamorphopsia, dyschromatopsia, micropsia and headaches occurring within hours of exposure are also commonly experienced. Fundus findings initially include a yellow-white spot in the fovea, sometimes changing to a red spot with a pigmented halo over several days. On fundus autofluorescence, solar retinopathy appears as hypo-autofluorescence at the fovea surrounded by an irregular ring of hyper-autofluorescence. OCT imaging typically shows disruption of the inner and/or outer segments of the photoreceptor layers, either with or without RPE defects. The literature indicates that defects of the inner photoreceptor segment are associated with worse VA than defects in either the outer segment or RPE. There may also be a transient increase in foveal reflectivity and reduced reflectivity of the RPE. There is no effective treatment for solar retinopathy. In many cases, visual acuity may recover to normal, however some patients have residual small central or paracentral scotomas. The extent of damage and prognosis are dependent on many factors including the duration and intensity of the light source during exposure, ocular pigmentation, ocular media clarity and environmental conditions.
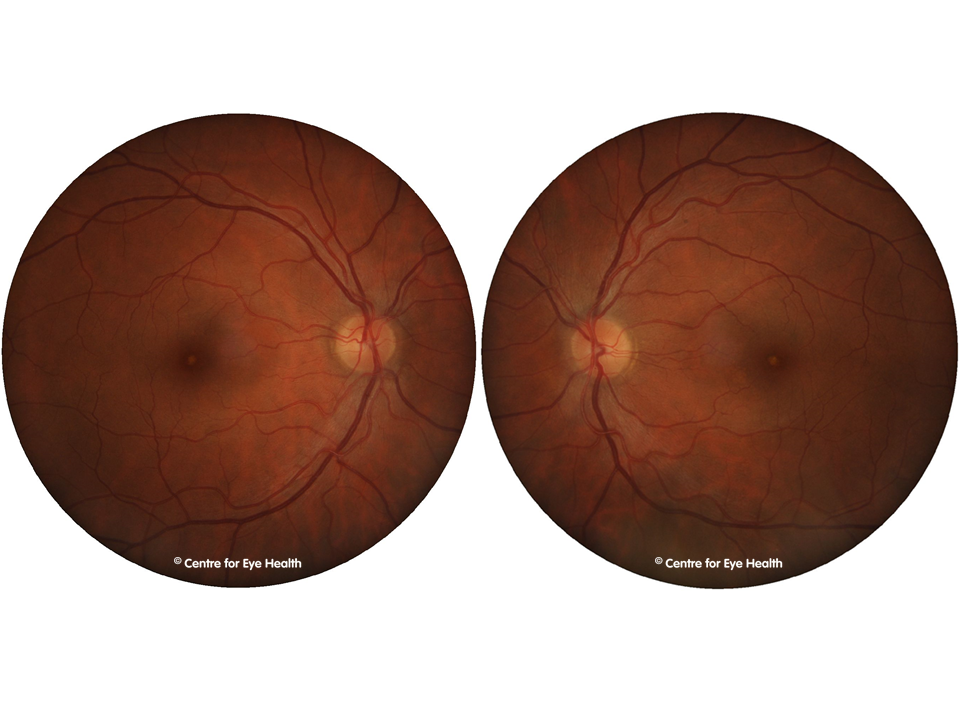)
CFEH Facebook Case #37
Case #37 PDF Download
For the original question click here
Answer
The patient’s fibrotic scar was most likely due to a large macular haemorrhage. Given the patient history of acute vision loss while undertaking heavy lifting, this was most likely a Valsalva retinopathy. Valsalva retinopathy is a preretinal haemorrhage that usually involves the macula. It is associated with a history of straining (eg coughing, vomiting, Valsalva manoeveur, weight lifting etc). It is thought that closure of the glottis causes an acute rise in intrathoracic or intraabdominal and associated rapid increase in intraocular venous pressure, leading to capillary rupture. The macular haemorrhage in some cases may resolve without intervention, returning the vision to normal or near normal. In cases where dense premacular haemorrhages affect vision. Nd:YAG laser may be used to puncture a hole in the posterior hyaloid and drain the blood into the vitreous cavity. This procedure has several potential complications however, including retinal detachment, ERM and macular hole formation.
CFEH Facebook Case #36
Case #36 PDF Download
For the original question click here
Answer
The follow up OCT shows the presence of sub-retinal fluid, indicating this is likely to be neovascular (late) AMD. In this case, OCT imaging has allowed early detection of the change in the absence of any symptoms and minimal to no change in the fundus photographic image. Twelve months previously, this patient was assessed as having a single large (confluent) drusen in the right eye and pigmentary changes as well as medium and small drusen. The left eye had medium and small drusen. At this time according to the Beckman AMD classification, the patient had intermediate AMD in the right eye and early AMD in the left, giving an approximate risk of conversion to neovascular AMD of 12% within five years. This case illustrates the importance of using OCT imaging on all AMD patients. With many practices now equipped with OCT technology, CFEH has produced an AMD chairside reference guide to help optometrists in the interpretation of the scans produced. The reference can be downloaded free by clicking here. OCT imaging is also available free to all patients through CFEH. Click here to download a referral form.
CFEH Facebook Case #35
Case #35 PDF Download
For the original question click here
Answer
Pathological Myopia. The Optomap images show significant chorioretinal atrophy centrally, as well as pigment degeneration peripherally. The fundus auto-fluorescence imaging reveals multiple zones of hypo-fluorescence – around and inferior to the optic disc, inferior to the macula and extensively throughout the periphery. The OCT shows a posterior staphyloma (an elongation of the globe resulting from scleral thinning) and a mirror artefact – often seen in high myopia. These retinal changes are consistent with pathological (high/degenerative) myopia. Pathological myopia is the progressive and excessive elongation of the globe, causing secondary changes involving the sclera, retina, choroid, vitreous, macula and optic nerve head. It is defined as myopia with a spherical equivalent refractive error of -6.00 DS or greater or an axial length above 26mm. The following are all conditions that may be associated with pathological myopia:
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
For a detailed discussion of pathological myopia, log in to Learning for Vision if you are a member or create a new account then click here.
CFEH Facebook Case #34
Case #34 PDF Download
For the original question click here
Answer
Differentials include: iris leiomyoma, iris melanoma, iris naevus and a metastases from a primary tumour elsewhere. The reported stability of the lesion makes it less likely to be a melanoma or metastases, however a final diagnosis can only be made following a biopsy. Imaging shows the lesion to be a raised, amelanotic lesion with defined margins and a lobular vascularised surface at the nasal peripheral iris of the left eye. Anterior OCT shows shows no invasion of the lesion into the angle and shows the anterior surface to be hyper-reflective. An iris amelanotic melanoma is a non-pigmented, benign nodular tumour that is typically found on the inferior iris, usually associated with superficial blood vessels. They are usually at least 3mm in diameter and 1mm thick and exhibit slow growth with extension across the iris surface. This expansion gives rise to the possibility that the angle and anterior ciliary body may be infiltrated. Thus, complications of amelanotic iris melanomas can include pupillary distortion, ectropian uveae, hyphema, cataract and glaucoma. An iris leiomyoma is an extremely rare benign tumour with an appearance similar to an amelanotic melanoma. Iris leiomyomas are tumours that originate from the smooth muscle, typically this is the sphincter muscle but less commonly can also be the dilator muscle. The mass is typically vascularised, either flat or slightly elevated and usually remains stationary over many years. They are usually found at the region of the sphincter muscle, although rarely can also be found in the iris periphery and anterior chamber angle. Leiomyomas can appear either non-pigmented, lightly pigmented or transparent (greyish white or pinkish). Complications may rarely include secondary glaucoma and decreased vision if the leiomyoma affects the visual axis. This lesion needs to be referred to an ophthalmologist for a likely biopsy.
CFEH Facebook Case #33
Case #33 PDF Download
For the original question click here
Answer
OCT-Angiography at CFEH OCTA is a non-invasive, high-resolution imaging method of visualising microvasculature by detecting motion contrast from flowing blood (without the injection of dye). This emerging technology has great potential for clinical practice as a quick, low risk means of assessing retinal vasculature. Conditions where OCTA may provide significant benefit include:
- Inner retina disease (retinopathies, vascular acquired or congenital disorders and malformations such as
- Epiretinal membranes
- Coat’s disease
- MacTel
- Macroaneurysm
- Diabetic retinopathy
- Cotton wool spots
- Retinal vein occlusion
- Outer retinal or choroidal disease (choroidal neovascularisation, age related macular degeneration)
- Disc perfusion abnormalities (eg glaucoma, ischaemic optic neuropathy, optic neuritis)
- Anterior eye vascular anomalies (eg iris and corneal neovascularisation)
- Pigmented lesions
The Centre for Eye Health utilises this new technology where appropriate for a greater understanding of the disease process in suitable patients. To refer a patient for OCTA imaging, please download the referral form here.
CFEH Facebook Case #32
Case #32 PDF Download
For the original question click here
Answer
Birdshot chorioretinopathy (later stage). Retinal photography and Optomap imaging showed yellow-orange spots deep to the retina scattered throughout the posterior pole and nasal to the disc. Fundus autofluorescence imaging shows mild hyperfluorescence of the lesions. Spectralis OCT imaging of the lesions show increased signal transmission through to the choroid, suggesting mild RPE thinning in these areas. There is mild preretinal fibrosis inferior to the macula. This clinical picture appears consistent with the diagnosis of birdshot chorioretinopathy. Birdshot chorioretinopathy is a rare disorder that falls under the umbrella of conditions known as white dot syndrome. White dot syndromes are thought to manifest from inflammation of the choriocapillaris and RPE. It most commonly affects women aged between 40 and 70 and can be associated with infections such as cytomegalovirus, cat scratch disease and Lyme disease, as well as vasculitis and uveitis. Some association has been made with birdshot chorioretinopathy and the HLA-A29 gene, and it has been postulated that the condition may result from a compromised immune system. The condition typically exhibits multiple cream-coloured oval lesions that appear to radiate from the optic disc. Patients can experience blurred vision, poor night vision, increased light sensitivity, paracentral scotomas, floaters and problems with colour vision.
CFEH Facebook Case #31
Case #31 PDF Download
For the original question click here
Answer
1. Amelanotic choroidal naevus Choroidal naevi are common, benign findings, typically found on routine fundus examination. A minority of these lesions, such as this one, are “amelanotic” as they do not display the typical greyish colouring. The presence of overlying drusen is an indication of chronicity of the lesion and thus a reduced risk of progression. The risk factors for malignant transformation of a naevus include the following: o Thickness > 2mm o Presence of subretinal fluid o Lipofuscin (orange pigment) overlying the lesion o Presence of symptoms o Location < 3mm of the foveola or at/near the optic disc In this case, a review cycle (including fundus photography and OCT) at 3 months, 6 months, then annually once stability is confirmed, is recommended due to the proximity to the foveolar. 2. Reticular pseudodrusen These intriguing lesions, also known as subretinal drusenoid deposits, were first described in the literature relatively recently (in 1990) and carry a high association with late AMD. They have featured in a previous Learning for Vision condition spotlight. Click here for more information.
CFEH Facebook Case #30
Case #30 PDF Download
For the original question click here
Answer
This condition was covered a couple of months ago in our Facebook case presentations (case 15). However, this is a vastly different and more advanced case. Familial dominant drusen is typified by a bilateral, relatively symmetrical distribution of drusen, most prominent temporal to the macula, but also seen outside the vascular arcades and nasal to the optic nerve head. The peripheral retina normally remains free of drusen. Historically, there were two conditions defined in the early literature as separate entities: Doyne’s honeycomb dystrophy described a presentation similar to that seen in this patient, while Malattia Leventinese specifically referred to drusen radiating temporally from the fovea. Further studies later revealed that the two conditions both occur due to a single defect in the gene that codes for the protein fibrillin 3, and were in fact different expressions of the same disease. As both this and case 15 illustrate, the phenotype is extremely variable. This patient has an advanced form of the disease with large, coalescent drusen, RPE irregularities and disruption of the outer retinal layers. Some patients with this condition may develop choroidal neovascularisation, however in this case there was no obvious evidence of current exudative changes.
CFEH Facebook Case #29
Case #29 PDF Download
For the original question click here
Answer
The lesions are most likely peripheral choroidal neovascularization (CNV) – there are 2 associated haemorrhages and a fibrous component to the choroidal lesion, suggesting this diagnosis. This type of choroidal neovascularization results in an increased risk of macular CNV as well as sub-retinal haemorrhages and can be associated with polypoidal choroidal vasculopathy (PCV). The incidence of PCV is high in African Americans, moderately high in Asians, and relatively low in Caucasians. It is a chronic vascular abnormality of choroidal blood vessels with persistent serous leakage and recurrent bleeding. The usual age of onset is between 60 and 70 years. It is characterised by branching choroidal vessels with aneurysmal dilations, which appear as red/orange structures in the macular and/or peripapillary areas. These lesions can be seen in some eyes during fundoscopy, but are more easily visualised with fluorescein angiography (FA) and indocyanine green angiography (ICGA). PCV lesions in the peripheral fundus such as in this case have also been reported but are less common. Although the lesion in this case is unlikely to require treatment, referral to a retinal specialist is still necessary.
CFEH Facebook Case #28
Case #28 PDF Download
For the original question click here
Answer
Bergmeisters papilla – a persistent remnant of the hyaloid artery During gestation the hyaloid artery nourishes the developing crystalline lens. The artery is surrounded by a perivascular sheath and before birth this artery regresses, however this regression is sometimes incomplete. Remnants of this artery can be seen in 95% of premature infants and 3% of full term infants. The term Bergmeisters papillae can be used to describe persistence of either the central vascular core (as in this case) or of the fibroglial sheath. The latter appears as a tuft of glial tissue that is most commonly located on the nasal aspect of the disc – although this can be highly variable. This finding is of minimal clinical significance.
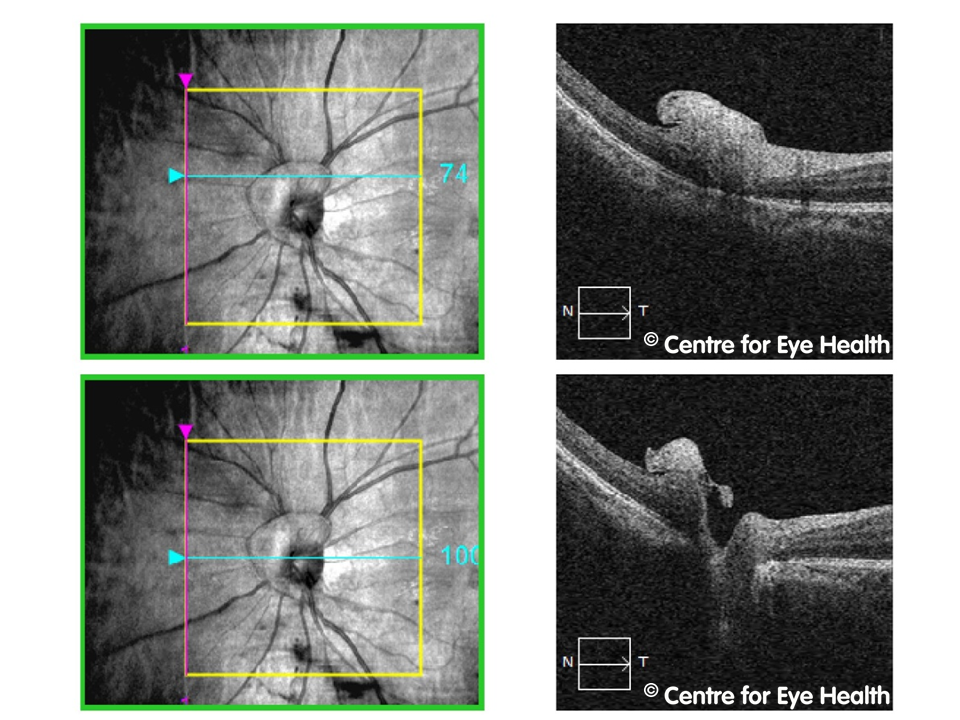)
CFEH Facebook Case #27
Case #27 PDF Download
For the original question click here
Answer
Melanocytoma of the optic nerve head This patient has an irregularly pigmented dark lesion that is located partly on the disc, extending over the superior margin and onto the retina. The disc margins appear somewhat undefined. The OCT line scans show a dome-shaped elevation at the superior aspect of the disc, with posterior optical shadowing. The b-scan ultrasound shows a small area of hyper-reflectivity at the right optic nerve head, indicating an elevated and probably solid lesion. A melanocytoma of the optic disc is a typically benign pigmented tumour. It is a variant of a naevus and can also involve the uveal tract (iris, ciliary body and choroid). They usually present as an isolated entity, with no proved systemic associations, and rarely cause visual impairment, however associated complications may occasionally occur. These complications can include disc oedema, intraretinal oedema, subretinal fluid, retinal haemorrhage and exudation, central retinal vein occlusions, neuroretinitis and malignant transformation (1-2% of cases).Melanocytoma has equal incidence in all races with a slight predilection for females. It is typically unilateral although bilateral cases have been reported in children. Lesions are typically small with average measurements of 2mm in diameter and 1mm in apical thickness. Recommended management involves 6-12 monthly review (depending on the level of risk or concern over malignancy). Examination should include stereoscopic examination, photography, OCT and automated perimetry.
CFEH Facebook Case #26
Case #26 PDF Download
For the original question click here
Answer
Right eye – glaucoma suspect. Left eye – (pre-perimetric) primary open angle glaucoma The small, tilted and obliquely inserted discs complicate a glaucoma assessment. Similarly high myopia and associated staphylomas can also lead to variable imaging and visual field results, in particular with respect to normative comparisons. As a result, the complete clinical picture needs to be assessed to arrive at an appropriate diagnosis. Pigment dispersion syndrome (PDS) is usually characterised by two or more of: iris transillumination, pigment deposition on the central corneal endothelium and increased pigmentation of the trabecular meshwork (Balidis et al., 2002). In this case, a diagnosis of PDS is not appropriate given the sparse nature of endothelial pigment deposition in conjunction with a lack of transillumination defects. There is no evidence of glaucomatous thinning of the NRR or OCT RNFL and GCA in the right eye. In conjunction with variable visual fields results showing no repeatable loss, the right eye was classified as a glaucoma suspect. The left NRR was thin inferiorly with stereo assessment with a correlating relative thinning of the OCT RNFL and GCA analysis (despite being classified as within the normal range). Given that the IOP was elevated but there was no repeatable field defect, the left eye was classified as having pre-perimetric primary open angle glaucoma. A prostaglandin analogue was prescribed for the left eye only, lowering the IOP to 15mmHg. The patient is now undergoing collaborative care with the referring optometrist and CFEH’s Glaucoma Management Clinic.
CFEH Facebook Case #25
Case #25 PDF Download
For the original question click here
Answer
Radial retroiridal pigmented lines The pigment originates from constant abrasion of the posterior layers of the iris with the anterior surface of the lens. The release of pigment is possibly associated with age-related changes of the iris pigmented epithelium being decrease in melanin and vacuolization as well as necrosis of the pupillary border. Radial retroiridal pigmentation was previously thought to be remnants of the tunica vasculosa retroiridalis however observations of the sign mainly in adults and not children did not support this theory. Clinical features: Radial retroiridal pigmented lines can occur in one or both halves of the lens periphery and on occasions the whole circumference of the lens, usually in the lower nasal quadrant. The central pupillary area is commonly absent of pigment. Radial pigmentation is usually seen in patients after the 5th decade of life. Clinical implications: Radial retroiridal pigmented lines were more likely seen in hyperopes which may be associated with crowding of the anterior segment structures causing more abrasion of the iris on the lens. Given that radial pigmentation is a possible risk factor for development of glaucoma in particular related to pigment dispersion, a glaucoma work-up including gonioscopy, applanation tonometry should be performed and these patients closely monitored. Systemic Associations: It has also been suggested that these pigmented lines can be associated with diabetes and arteriosclerosis.
CFEH Facebook Case #24
Case #24 PDF Download
For the original question click here
Answer
Leber’s Hereditary Optic Neuropathy. LHON usually affects men with onset between ages 10 and 30 years of age. There are often no symptoms until an acute, severe painless loss of vision is experienced with acuity less than 6/60 and central or cecocentral field loss. This loss of vision is usually permanent, however a small percentage (10-20%) do recover some vision. Approximately a quarter of all cases are bilateral at onset, however unilateral cases will typically also start to show symptoms in the other eye 2-3 months after the first eye becomes symptomatic. The fundus can either appear entirely normal (20% cases), or can show pseudopapilloedema, peripapillary telangiectasia and tortuosity of retinal arterioles. Fundus findings may be subtle but can preceed vision loss. Atrophy of the optic discs can be seen within 6 weeks of the acute vision loss, a finding that is accompanied by a dense central scotoma. Pathologically, there is damage to the retinal ganglion cells, axonal degeneration and demyelination then atrophy from the optic nerve back to the Lateral Geniculate Nucleus. The RPE and photoreceptor layers are spared. LHON arises from a mutation in the mitochondrial DNA, affecting the genes that code for NADH Dehydrogenase protein which is required for normal mitochondrial phosphorylation. The mutation is inherited only from the mother. Non-symptomatic carriers of the gene mutation have been found to show a temporal thickening of the retinal nerve fibre layer (Savini et al. 2005), and may also show a subtle impairment of optic nerve function, including reduced contrast sensitivity, altered colour vision and subnormal electrophysiology results (Sadun et al. 2006) No treatment has been shown to be effective, although new medical and gene therapies remain under investigation. Genetic counselling and referral to low vision services are recommended.
CFEH Facebook Case #23
Case #23 PDF Download
For the original question click here
Answer
Lattice Degeneration with a sub-clinical retinal detachment. The Optomap image shows an oval, white/gray band at the inferior temporal equator. The orientation of the lattice is parallel to the ora serrata. The white lines crossing the lattice represent sclerosed blood vessels. Over time, lattice degeneration shows increased pigmentation and retinal thinning, possibly resulting in retinal holes. OCT imaging of this band of lattice shows thinning of the retina (this is seen in the horizontal line scan, by comparing the retina at the left side to the right side of the image). There is also vitreoretinal traction resulting from the strong adhesion of the vitreous to the lattice. This has resulted in a retinoschisis (splitting of the retina) as can be seen most clearly in the horizontal OCT line scan, and also a sub-clinical retinal detachment where the sensory retina has separated from the RPE. This detachment can be seen in the right hand side of the vertical line scan. Other findings evident on the Optomap image include a posterior vitreous detachment (as evidenced by the dark floater seen below the optic disc) and spots of chorioretinal atrophy temporal to the area of lattice degeneration. Lattice degeneration is found in 6-10% of the normal population. It first appears in young patients with a peak incidence in 2nd-3rd decade of life and is bilateral in up to half of all cases. It is most commonly found in the superior and temporal retinal periphery and is more commonly seen in myopes. In an acute PVD, tears may develop along the posterior edge of the lattice. This patient was referred to a retinal specialist for consideration of prophylactic treatment. For patients with lattice and no associated detachment or tear, an annual dilated fundus examination is recommended with advice to return urgently if any new signs or symptoms occur. Scleral indentation and OCT imaging is highly recommended as these can detect any anomalies such as the sub-clinical detachment seen in this patient.
CFEH Facebook Case #22
Case #22 PDF Download
For the original question click here
Answer
A coloboma. A typical coloboma forms as a result of the embryonic fissure failing to close completely during development. During normal development, the fusion of the embryonic fissure begins at around 33-40 days post conception. A coloboma occurs when there is a fault in this closure process, with the location of the anomaly determined by the location of the closure fault. The size of the coloboma can be variable depending on how much of the fissure fails to fuse. Typically colobomas are found inferior-nasally. Colobomas can occur in the retina/choroid, the optic nerve and/or the iris and if a coloboma is found in one location, a complete examination should be performed as it may also be present in additional locations. Optic disc colobomas cause the disc to appear enlarged and excavated, often more so in the vertical meridian. Location is usually at the inferior aspect of the disc so the inferior neuro-retinal rim is typically either thin or absent, and normal disc tissue is confined to a small superior wedge. Visual acuity is often decreased and there is an associated superior scotoma. In this case, the patient has a large coloboma in both eyes. The optic disc is completely involved in the right eye and partially in the left. The excavation of both can be appreciated by looking at the B-scan ultrasound. There was an associated iris coloboma inferiorly in the right eye. The left eye still had some macula function as the edge of the coloboma is adjacent to the macula rather than encompassing it as it does in the right eye (fig 2 below). Colobomas may show either autosomal dominant or autosomal recessive inheritance or may be an isolated anomaly. Another cause may be teratogens – agents that disturb the normal development of an embryo or foetus. These include exposure to thalidomide in the first trimester and possibly LSD ingestion, although the evidence for this is not conclusive. Various other genetic disorders also have coloboma as a rare manifestation, however the correlation is, again, lacking in conclusive evidence. For more information on this condition and other neuro retinal rim anomalies, a video lecture on this topic is available through Learning for Vision – click here
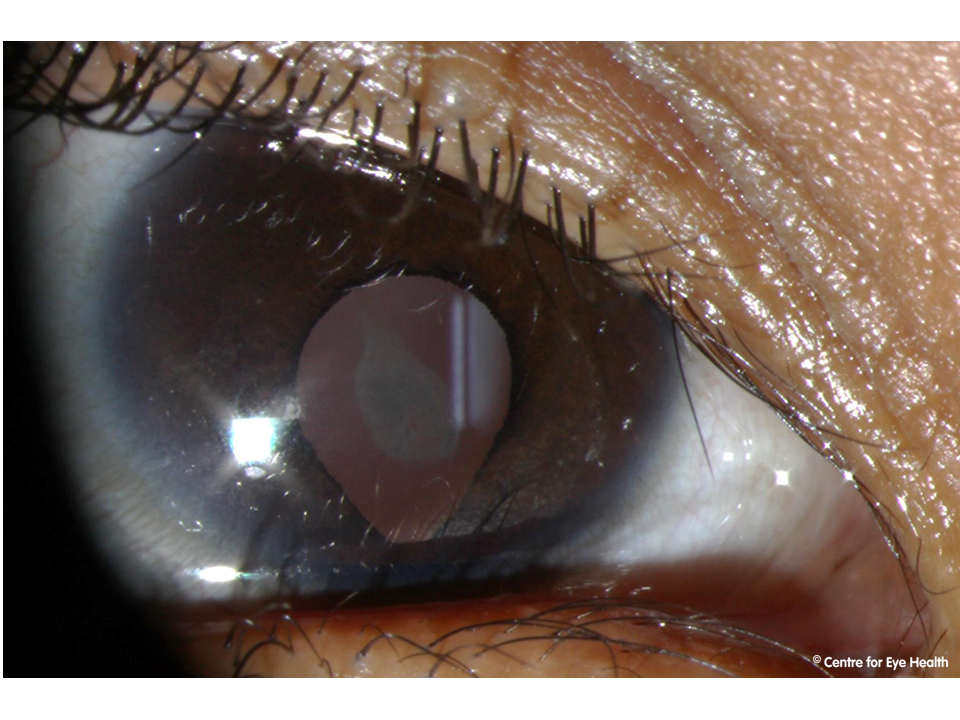)
CFEH Facebook Case #21
Case #21 PDF Download
For the original question click here
Answer
Ocular Albinism The anterior eye image shows transillumination defects while the Optomap image suggests reduced retinal pigmentation and the OCT line scans reveal foveal hypoplasia (an absence of foveal reflex and foveal pit). These findings, and the reduced visual acuities suggest a diagnosis of ocular albinism. Foveal hypoplasia is a common to all types of ocular albinism, however this can be variable in its magnitude and is important in determining visual acuity. The degree of hypopigmentation varies according to the type of albinism. Other associated signs and symptoms can include reduced visual acuity, photophobia, congenital nystagmus, refractive errors, prominent choroidal vasculature, optic disc hypoplasia and transillumination defects. Ocular albinism usually shows an x-linked inheritance pattern, although occasionally can be autosomal recessive. Female carriers of the x-linked type can show partial iris translucency and scattered areas of depigmentation but are otherwise asymptomatic. As part of the optometric management, an optic nerve head assessment, gonioscopy, visual fields and IOPs should be performed to exclude pigment dispersion syndrome. Management options for ocular albinism include low vision aids, tinted lenses and consideration of genetic counselling.
CFEH Facebook Case #20
Case #20 PDF Download
For the original question click here
Answer
Punctate inner choroidopathy (PIC) PIC is a relatively uncommon inflammatory condition that is one of the “white dot syndromes’. Characteristic fundus findings include multiple yellow-white lesions at the level of the choroid and RPE, usually limited to the posterior pole, which leave atrophic pigmented scars on resolution. Initial presentation is usually associated with a loss of central visual acuity, photopsia and scotoma. PIC predominantly affects young myopic females. The visual prognosis for most patients is good, however for a small percentage of patients there is a moderate risk of developing irreversible vision loss due to associated choroidal neovascularization (CNV) or sub-retinal fibrosis. These usually occur within 12 months of initial presentation. In the absence of symptoms, regular optometric reviews, Amsler grid home monitoring and periodic OCT imaging is required to monitor the condition and allow detection of vision threatening CNV.
CFEH Facebook Case #19
Case #19 PDF Download
For the original question click here
Answer
Unilateral chorioretinal folds The horizontal stripes in the left fundus image are suggestive of chorioretinal folds. These are parallel grooves in the inner choroid, also normally involving Bruch’s membrane the RPE and possibly the retina. A vertical line scan through the left fundus clearly demonstrates the nature of these folds. Chorioretinal folds tend to appear as yellow streaks in the fundus (due to the stretching of the RPE) alternating with darker streaks (RPE compressions). By contrast, retinal folds not involving the choroid are more likely to present with streaks of a finer appearance. The aetiology of chorioretinal folds is varied. Idiopathic chorioretinal folds are usually asymptomatic with a bilateral presentation and they are most often found in hyperopes. Other more sinister causes can include ocular hypotony, retrobulbar tumours, choroidal tumours, choroidal neovascularization and papilloedoma. Hence, a new discovery of chorioretinal folds requires further investigation (often with fluorescein angiography and/or neurological scans) to exclude any pathology within or behind the eye. Patient symptoms such as metamorphopsia would suggest this to be a newly formed condition, increasing the urgency of these investigations.
CFEH Facebook Case #18
Case #18 PDF Download
For the original question click here
Answer
The slit lamp images show fine, superficial vascularization extending from the limbus, irregular yellow lipid deposits and corneal opacification/scarring. Anterior OCT images (below) show thinning of the cornea with a thickness measurement in the lower scan of just 213µm
Terrien’s marginal degeneration typically shows superior thinning of the cornea with associated lipid deposit and neovascularization. Those with this condition often have reduced vision associated with against the rule astigmatism. The corneal epithelium remains intact in this condition, so the area will not stain with fluorescein.
There are two types of presentation. The first is a non-inlfammatory type that typically presents in males over 40 years of age and has a slow rate of progression. The second is associated with episcleral and scleral inflammation and presents in patients aged 20-30 years of age. This second type has a more guarded prognosis with patients at risk for corneal perforation (particularly following trauma) which would require lamellar keratoplasty. Considering this risk, the patient was referred to ophthalmology.
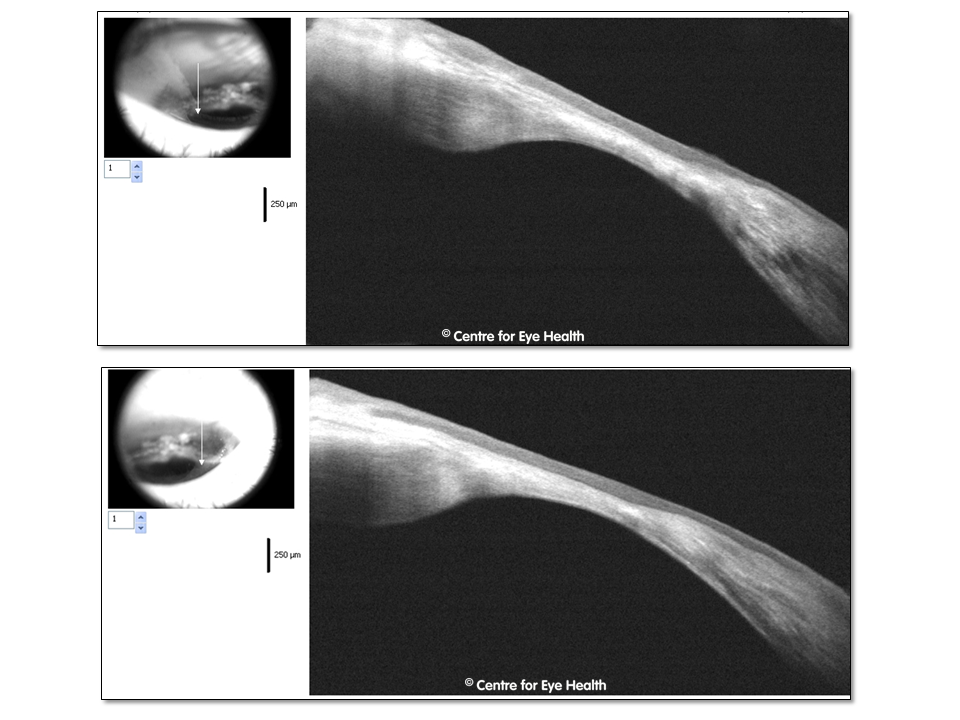)
CFEH Facebook Case #17
Case #17 PDF Download
For the original question click here
Answer
Pigmented Paravenous Chorioretinal Atrophy (PPCRA)
This is a bilateral, progressive condition that is characterised by pigmentary changes including RPE atrophy and pigment clumping around the blood vessels. The OCT images show a reduction in the outer nuclear layer thickness, as well as disappearance of the Ise and interdigitation zone. The hyper-reflective areas seen in the OCT images within the retina are due to RPE migration and increased reflectance seen in areas of the choroid is due to the drop out of RPE cells.
The exact aetiology of this condition is unknown, and speculation exists that it may be either genetic in nature, developmental or an immune response due to associated inflammatory conditions.
Sufferers often remain asymptomatic and the threat to vision is not high as the condition is usually stable. Recommended management is to review the fundus appearance and visual fields annually to document progression. An ERG can help with diagnosis and monitoring of this condition. In this case, both an ERG and EOG were performed at CFEH with the ERG showing a normal scotopic and reduced photopic response. The EOG showed an abnormal Arden ratio. Given the presence of significant field defect, Estermann field assessment may be required to assess suitability to drive.
This case and a series of other unusual eye conditions are presented in a video lecture “Case Studies on Unusual Eye Conditions”, available for viewing through Learning for Vision and approved for 2 therapeutic CPD points. Go to www.learningforvision.com.au to purchase this and other lectures from our catalogue.
CFEH Facebook Case #16
Case #16 PDF Download
For the original question click here
Answer
Although we find, as expected, a superior arcuate-type defect in the right eye which exhibits structure-function correlation with the inferior NRR (seen in stereo), RNFL and GCIPL loss, the left eye result is surprisingly clear. This is in spite of structural findings that are not too dissimilar between the eyes (for example, the RNFL symmetry index was at 84%; comparison of raw GCIPL thickness values between the eyes in fact shows the left eye to be slightly more depressed). Poor structure-function relationship has been noted in a high number of cases of early glaucoma, whereby some patients show obvious signs of structural loss with absent visual field defects (i.e. pre-perimetric glaucoma). The visual field results of such patients have been found to be different to that of a completely healthy patient, however the results are all generally “green”, and show no failure of any typical visual field criteria for defect (e.g. MD, PSD, event analysis)
)
CFEH Facebook Case #15
Case #15 PDF Download
For the original question click here
Answer
Familial Dominant Drusen Retinal photography and the optomap image show diffuse drusen throughout the posterior pole, nasal to the disc, affecting the macula. Fundus autofluorescence shows a pattern of speckled hyper and hypo-autofluorescence. The OCT line scans show drusenoid elevations of the RPE. This clinical picture suggests the diagnosis of familial dominant drusen, also known as Malattia leventinese, Doyne honeycomb retinal dystrophy or autosomal dominant radial drusen. It is unclear if these names represent one disorder or a spectrum of disorders. Malattia leventinese has been specifically defined as familial dominant drusen when the drusen are centred on the fovea in a radiating pattern. Familial dominant drusen typically present in the 2nd-3rd decade of life with patients remaining asymptomatic until the 4th decade of life when metamorphopsia and/or decreased vision can begin. The condition is typified by bilateral, relatively symmetrical drusen that are prominent on the temporal side, but also seen outside the arcades and nasal to the optic nerve. The peripheral retina normally remains free of drusen. Diagnosis is confirmed if the patient reports family members with a similar pattern of drusen. As the condition progresses drusen become confluent and pigment hyperplasia develops. Subsequent geographic atrophy and choroidal neovascularization can cause significant vision loss. Patients should self-monitor their vision at home with an amsler grid and undertake regular review, including OCT imaging. If choroidal neovascularization develops, referral to an ophthalmologist for review is required. A 2012 study using a small sample size found treatment with Argon green laser may have a beneficial effect on patients with familial dominant drusen, however further assessment and follow up is still needed.
CFEH Facebook Case #14
Case #14 PDF Download
For the original question click here
Answer
Intrachoroidal cavitation. The fundus images show a pale, well circumscribed circular lesion inferior to the fovea with irregular pigmentation, a circumscribed area of hypo auto-fluorescence and an associated wedge defect. There was a corresponding superior paracentral field defect. Spectralis OCT through the lesion shows loss of the neurosensory retina and retinal pigment epithelium (RPE) overlying a posteriorly bowed sclera and cavities within the choroid. Intra-choroidal cavitation has been described as a focal loss of the outer retina and RPE, with the remaining inner retina making direct contact with the sclera resulting in a “pseudo-fovea” appearance. Fluorescein angiography shows mild hyperfluorescence in the early phases and mild hyperfluorescence in late phases. Early studies have found intra-choroidal cavitations to be more common in older subjects and those with longer axial lengths. They occur most frequently in the lower temporal quadrant and have a significant association with retinoschisis. The sclera is bowed posteriorly in just over 50% of cases. Further studies are required to investigate these associations and long term sequelae.
CFEH Facebook Case #13
Case #13 PDF Download
For the original question click here
Answer
Choroidal neovascularization (CNV) associated with chronic Central Serous Chorioretinopathy (CSCR) Retinal photography and more notably the OCT Scanning Laser Ophthalmoscope image from the initial visit, showed scattered RPE disruptions in the left eye. Baseline OCT line scans showed corresponding RPE and photoreceptor layer (ONL and EZ) disruption with migration of the RPE. This clinical appearance, combined with the patient profile and subsequent autofluorescence image, is consistent with CSCR which falls into the spectrum of pachychoroid spectrum disease (for more information on this, click here.) The current images showed exudates, subretinal fluid and intraretinal cystic spaces with a significant increase in superior macular thickness compared to baseline. Fluorescein angiography or possibly OCT Angiography would confirm the diagnosis, however these findings are consistent with CNV formation. CSCR is the second most common cause of CNV following AMD. Visual function loss can also occur if serous detachment of the photoreceptors persists for over 3 months. As a result, while most cases of CSCR are self-limiting, chronic or recurrent cases warrant referral to a retinal ophthalmologist
CFEH Facebook Case #12
Case #12 PDF Download
For the original question click here
Answer
Polypoidal choroidal vasculopathy. Note that the round/yellow white lesion on the inferotemporal macula is a scar due to previous trauma to the right eye. Polypoidal choroidal vasculopathy (PCV) is a chronic vascular abnormality of choroidal blood vessels with persistent serous leakage and recurrent bleeding. It is characterised by branching choroidal vessels ending in polyp-like aneurysmal bulges. These lesions usually appear in the macula and/or near the optic disc. There are often associated pigment epithelial detachments (PEDs). PCV has a similar clinical appearance to neovascular AMD, however PCV is not associated with drusen and tends to progress at a slower rate than neovascular AMD. The usual age of onset is 50-65 years with the highest incidence seen in African Americans, followed by Asians and with a low incidence in Caucasians. A definitive diagnosis of PCV requires indocyanine green angiography. Fundus autofluorescence imaging provides a non-invasive aid in identifying the sites of polypoidal lesions, which appear hypo-autofluorescent. OCT can help in detection of associated abnormalities such as PEDs. Multiple haemorrhagic PEDs were observed with OCT in this case. Polypoidal lesions can resolve spontaneously and be replaced by subretinal fibrosis, RPE hyperplasia and/or atrophy. Lesions may also lead to choroidal neovascularisation. Generally, a conservative approach to treatment is recommended unless there is persistent or progressive exudation that is macula-threatening. Treatment options include thermal laser photocoagulation (limited use in sub-foveal lesions), photodynamic therapy and anti VEFG agents to treat leaking aneurysms or polypoidal components within the vascular lesion. Most recently, a combination of PDT and anti-VEGF agents has been favoured due to a possible synergistic effect.
CFEH Facebook Case #11
Case #11 PDF Download
For the original question click here
Answer
Torpedo maculopathy with corresponding Amsler grid distortion and visual field loss inferonasally. Torpedo maculopathy is a congenital, solitary, chorioretinal lesion. There are no known associated congenital, systemic or ocular abnormalities. Distinctive features of torpedo maculopathy include a location temporal to the fovea, hypopigmentation and a horizontally oval, spindle-shaped lesion resembling a torpedo. This lesion typically has a pointed “head” directed toward the fovea and a round or frayed tail at the temporal margin. Visual acuity is generally not affected as the lesion does not involve the central fovea. As in this case, visual fields testing with 10-2 threshold perimetry showed a corresponding inferonasal defect. Amsler grid can be used as a screening assessment however it should be noted this may be less sensitive to smaller lesions. OCT imaging shows a neurosensory retinal detachment with a large cleft (posterior detachment of the RPE) and attenuation of the outer retinal layers including the inner segment ellipsoid line and RPE. The choroid is also more visible in the area of the lesion. Fundus autofluorescence imaging of the lesion reveals a predominantly dense hypo-autofluorescence pattern with mottled areas of iso- or hyper-autofluorescence. In terms of management for this case, although the lesion is congenital in nature and not known to progress, it is still important to establish stability and monitor progression particularly considering its proximity to the fovea. Therefore, routine review with photographic documentation for comparison and repeat imaging with OCT is recommended. It is also worthwhile for the patient to self-monitor their symptoms with the Amsler grid and patients should be advised to seek medical attention should any new or change in symptoms arise.
CFEH Facebook Case #10
Case #10 PDF Download
For the original question click here
Answer
Suspicious iris naevus. Iris naevi are relatively common pigmented lesions on the iris. Though generally innocuous, they may rarely convert to iris melanomas and therefore must be examined and monitored carefully. Shields et al, 2013 devised an easy mnemonic to recall features associated with higher risk of conversion to malignancy: A-Age (presentation<40 years) B-Blood (hyphema) C- Clock hour-inferior location D-Diffuse configuration E-Ectropian Uveae F-Feathery margin Other features that may represent a risk factor for malignancy include any iris distortion, anterior chamber cells or flare, dilated episcleral vessels, vascularisation or focal cataract. It should also be noted that pigmented iris lesions greater than 3mm in diameter or greater than 1mm thickness increases the likelihood of a choroidal melanoma. Gonioscopy is important in the assessment of elevation or expansion of iris naevi and to exclude invasion of the angle in more peripheral presentations. In this case, gonioscopy shows ectropian uvea which is a condition where the iris pigmented epithelium curls around the margin of the pupil to reach the anterior surface of the pupil margin. This results in distortion of the pupillary margin as noted on the anterior eye photo and is one of the risk factors for malignanacy identified by Sheilds et al above. UBM can be used to differentiate a solid mass such as the one in this case from an iris cyst which is acoustically empty. Iris naevi appear as lowly reflective surface plaques such as is seen here. The thickness of the naevus can also be estimated, and in this case it is approximately 1mm thick. Additionally, the UBM image shows a ‘Lion’s paw’ appearance, which is a posterior extension of the iris, delimited by the lens. This appearance should raise suspicion of a possible nodular melanoma. Traditional OCT can be used in these cases to demonstrate thickening and iris contour however iris penetrance is relatively poor and therefore is less helpful in differential diagnosis compared to UBM or anterior specific OCT. This naevus exhibited the established risk factors of inferior clock hour location, ectropian uvea, iris distortion and a “lion’s paw” appearance on UBM. Due to these suspicious features, this patient was referred to an ophthalmologist for an opinion on management.
CFEH Facebook Case #9
Case #9 PDF Download
For the original question click here
Answer
Diagnosis: Small, congenitally tilted discs with parapapillary intrachoroidal cavitation (PICC) and a probable developing acquired juxta-papillary pit (indicated on the images below) with corresponding visual field loss. PICCs are a juxta-papillary or peripapillary detachment, characterised on OCT by a hypo-reflective space in the choroid that can have normal retina and RPE overlying it. They may be associated with glaucoma-like visual field changes and can appear as a yellowish round lesion inferior to the optic nerve. Those that don’t have this appearance only show the cavity posterior to the myopic conus. As well as congenital tilted discs, PICCs can affect those with pathological myopia and are most commonly found inferior to the optic disc. The Beijing eye study (2013) found PICC was present in 16.9% of highly myopic eyes (refractive error greater than 6 dioptres or axial length greater than 26.5mm) and were typically associated with highly tilted optic discs and the presence of a posterior staphyloma. 71% of eyes with PICC have been found to have glaucomatous visual field defects. In this case, it was difficult to determine whether there were co-existing glaucomatous changes. As a result, the Centre’s ophthalmologist’s recommendation was to review the patient in 6 months to check for any evidence of change or progression.
CFEH Facebook Case #8
Case #8 PDF Download
For the original question click here
Answer
Choroidal osteoma. Choroidal osteomas are rare, benign tumours of the choroid, usually affecting the juxtapapillary and macular areas. They are more prevalent in females and are usually unilateral (80% of cases). Age of onset can vary significantly, from a few months of age to late 60’s, but they are predominantly found in young adults. Osteomas are typically irregular in shape with sharply defined or scalloped borders, are slightly elevated and usually have fine vascular networks on the surface of the tumor. The colour of an osteoma is related to the level of RPE depigmentation which increases over time meaning that in early stages, they are orange-yellow in colour and in later stages more of a yellow-white colour. Looking at the clinical picture for this patient, we can see a juxtapapillary yellow-orange lesion that involves the macula. It is well demarcated with scalloped borders and there is associated RPE mottling at the macular. Fundus autofluorescence shows a speckled hyper- and hypo- autofluorescence pattern along the inferonasal and superior margins of the lesion. OCT images show an abrupt change in the choroidal architecture at the site of the lesion while overlying inner and outer retinal layers appear undisturbed. In this case, OCT imaging showed there was no apparent subretinal fluid, RPE/photreceptor alterations or subretinal haemorrhage, explaining why visual acuity is still so good. This patient was referred to a retinal specialist for further assessment and the diagnosis was confirmed by B-scan ultrasound which showed a strong acoustic shadow which is characteristic of choroidal osteomas. Although a choroidal osteoma is benign, most show slow growth over time and vision can be affected through several mechanisms:
Atrohpy of the RPE and choriocapillaris, producing a thin, yellow-grey area overlying the osteoma.
Choroidal neovascularization (CNV), the prevalence of which has been found by various studies to be between 31 and 47%. Tumors with overlying haemorrhage and an irregular surface are at highest risk of developing CNV.
Subretinal fluid, haemorrhage and serous retinal detachment due to a damaged RPE are often associated with osteomas in the absence of CNV. One long-term study found that 64% of eyes with sub-retinal fluid resolved spontaneously, but those that didn’t had poor visual outcomes. The 10-year probability of losing visual acuity to the level of 6/60 was found to be around 56%.
CFEH Facebook Case #7
Case #7 PDF Download
For the original question click here
Answer
Choroideraemia. Choroideraemia is a progressive degeneration of the RPE and choriocapillaris. It has an X-linked recessive inheritance pattern and is thus found mostly in males. Early in the disease process, the mid-peripheral photoreceptors are affected causing mottled areas of pigmentation as seen in this patient. Symptoms include problems with dark adaptation and night vision complaints. Presentation is usually in the first or second decade of life. Over time the areas of mottled pigmentation and atrophy progress producing a ring scotoma similar to retinitis pigmentosa (RP). This condition is distinguished from RP however by the marked choroidal and RPE atrophy that develops, the fact that the blood vessels are unaffected and the absence of optic atrophy. Although the onset of symptoms in this condition is in early childhood, most sufferers have a slow course of degeneration and progression with most maintaining good vision for 40-50 years. Looking at the imaging results from this patient, widespread choroidal and RPE atrophy can be seen in the mid-peripheral and peripheral fundus. There is associated pigment granularity in both eyes. The right eye shows an area of more advanced RPE and choroidal atrophy, seen on the inferonasal fundus. At this time there is a relative sparing of the central fundus in both eyes. OCT line scans show widespread irregularity in the inner segment ellipsoid line, inner RPE and outer retinal layers in the top and bottom images. The scan through the fovea however shows the central macula region to be relatively unaffected at this time. To confirm the diagnosis, electrophysiology testing was conducted at CFEH. Carriers of X-linked choroideraemia often show patches of subretinal mottled pigment and occasionally a lobular pattern of choriocapillaris and RPE loss, but are asymptomatic and have normal electrophysiology results. For this reason, the patient’s mother was also examined and her Optomap images are below: These images show reticular pigmentary changes in the peripheral fundus of both eyes which are hypo-autofluorescent as might be expected in a female carrier of choroideraemia. Due to the sensitive nature of the diagnosis, the patient was not immediately given a diagnosis but was referred to an ophthalmologist for confirmation first and referral for appropriate counselling services.
CFEH Facebook Case #6
Case #6 PDF Download
For the original question click here
Answer
Racemose Haemangioma. Racemose haemangioma is a rare, typically unilateral, non-progressive retinal vascular tumor. It is a malformation of the retinal vessels allowing direct connection between the arteries and veins. Clinically it presents as dilated and tortuous retinal vessels extending from the optic disc to the retinal periphery. The lesions, if very large, may occasionally result in intraretinal, vitreous or macular haemorrhage however fluorescein angiography usually shows stable, non-leaking lesions. Vaso-occlusive disease may also be associated with racemose haemangioma. Excluding these potential complications, there is typically no visual deterioration with this condition. Racemose Haemangioma can be associated with Wyburn-Mason Syndrome. This syndrome is a vascular condition typified by arteriovenous malformations of the face, orbit, retina and central nervous system. A retinal racemose hemangioma is often associated with a lesion in the ipsilateral midbrain in this syndrome and there is typically an increase in early mortality due to the tendency of the AV malformations to bleed causing stroke, neurological deficits and death. No treatment is available for retinal lesions, however intracranial lesions may be managed with surgery, radiotherapy or embolization. Once Wyburn-Mason Syndrome has been excluded, routine review is recommended.
CFEH Facebook Case #5
Case #5 PDF Download
For the original question click here
Answer
Pattern dystrophy. Funduscopy and retinal photography show both hyper and hypo pigmentary changes at both maculae. Fundus autofluorescence shows a pattern of hyper fluorescence at the macula. The OCT images show signs of RPE pigment migration (most notable in the middle image shown) and atrophy of the outer retinal layers, including the photoreceptors (most notable in the middle and lower images shown). The middle image also shows a hyperreflective sub-retinal lesion that is typical of a pattern dystrophy. Pattern dystrophy is characterised by the development of “patterns” of yellow, orange or grey pigment at the macula. It is typically a bilateral, autosomal dominant condition associated with mutations in the RDS/peripherin gene and has an age of onset ranging from 30-50 years. Presenting symptoms typically include a mild loss of best corrected visual acuity and metamorphopsia, both of which have been noted in this case, however some cases may remain asymptomatic. There are 5 main “pattern” presentations possible and these are categorised according to the distribution of the pigment deposits:
- Adult-onset foveomacular vitelliform dystrophy – is characterised by yellow, round, bilateral subfoveal lesions that typically have a central pigmented spot and may be slightly raised.
- Butterfly dystrophy (as in this case) – bilateral butterfly-shaped pigmentation at the level of the RPE. Usually there is an area of total RPE and photoreceptor layer loss.
- Reticular dystrophy – appears as a reticular network of pigmented lines criss-crossing over the posterior pole with pigmented “knots” at the intersection of these lines.
- Fundus pulverulentus – the rarest presentation, characterised by a coarse pigment mottling of the RPE in the macular area. This condition can be difficult to differentiate from other maculopathies.
- Multifocal dystrophy – characterised by irregular yellow-white flecks scattered throughout the fundus, similar to those seen in fundus flavimaculatus. Associated macular lesions can range from yellow/grey deposits through to areas of severe chorioretinal atrophy. Multifocal dystrophy does not have the “dark choroid” seen in fundus flavimaculatus on fluorescein angiography.
The prognosis depends on which type of Pattern dystrophy is present and the age of onset of symptoms. This patient has butterfly dystrophy and can be expected to maintain relatively normal acuity for much of his life, however atrophic lesions can develop with age, which would adversely affect this acuity. To download the CFEH Chair-side reference guides to help with your differential diagnosis of this and other macular condtions, please go to: http://www.centreforeyehealth.com.au/chairside-referrences/
CFEH Facebook Case #4
Case #4 PDF Download
For the original question click here
Answer
Fuchs uveitis syndrome (FUS) is a chronic, mild non-gramulomatous anterior uveitis. It has been estimated that 90% of cases are unilateral and patients can be either symptomatic or asymptomatic. There is a wide variety of possible presentations, however the classic signs are as follows:
- Hypochromia of the affected eye (iris heterochromia) – in this case the affected eye is the left eye which has notably less pigment than the right eye
- Fine keratic precipitates on the corneal endothelium – typically stellate or dendritic in appearance. These KP’s can be seen in the anterior photo given, and from the OCT we can see confirm that they are located on the endothelium. It should be noted that while mutton-fat KP’s may occasionally appear in FUS, they are more typically found in granulomatous uveitis or Posner-Schlossman Syndrome.
- Mild or low-grade iridocyclitis – this patient shows no signs of active inflammation at this time and reports no inflammatory symptoms in the past. The mild nature of the iridocyclitis can cause a patient to be asymptomatic as in this case.
- Iris atrophy – develops over time and is not yet apparent in this case.
Other characteristic features can include iris nodules (Koeppe nodules on the pupillary margin and Busacca nodules on the iris surface); prominent iris blood vessels (due to gradual iris atrophy); iris crystals (Russell bodies) and mild vitritis. IOP of the affected eye can also be elevated during an episode of iritis. Possible complications include:
- Sub-capsular cataract – caused by severe anterior inflammation (anterior sub-capsular opacity) or vitritis (cortical and posterior sub-capsular opacities)
- IOP elevation and secondary open angle glaucoma (SOAG) due to damaged aqueous outflow caused by recurrent episodes of uveitis.
Treatment of SOAG usually involves aqueous suppressants (β-blockers, α-agonists and carbonic anhydrase inhibitors). The use of prostaglandin analogs is controversial as it is thought they may exacerbate the inflammatory response or cause recurrent uveitis, and laser treatment may be contra-indicated in FUS. For more detailed information on this condition and its atypical variants, please refer to the article published by CFEH: Phu J, Trinh WH, Chau-Vo JS, Nivison-Smith L, Kalloniatis M. Atypical Features of Fuchs Uveitis Syndrome Optometry and Vision Science (2015) Vol 92 no 11 pp 394-403
CFEH Facebook Case #3
Case #3 PDF Download
For the original question click here
Answer
X-linked Juvenile Retinoschisis. Visual acuity can range from 6/6 to light perception. VA typically reduces during first 2 decades with a then slow decline until the 5th and 6th decades. Foveal changes are seen in all cases and peripheral schisis in approximately half. Funduscopically, the macula often shows folds radiating out from the foveolar. The macula changes associated with x-linked juvenile retinoschisis are best viewed with OCT. This patient’s images show characteristic cystic spaces within the retina in the inner nuclear layer and the outer nuclear/outer plexiform layer which is typical of this condition. There is a marked elevation of the retina adjacent to the fovea extending to the inferior retina known as a vitreous veil. These can lead to retinal detachment and vitreous haemorrhage. A defect in the XLRS1 gene is responsible for this condition. This gene encodes an amino acid protein called retinoschisin which mediates the interactions and adhesions between the photoreceptor, bipolar and Muller cells. Review by a retinal specialist is advised with electrophysiology testing sometimes useful in confirmation of the diagnosis. Prophylactic treatment of the retinoschisis is not generally advised as there is a high risk of retinal detachment, however genetic counselling and low vision aids / management is recommended.
CFEH Facebook Case #2
Case #2 PDF Download
For the original question click here
Answer
The lesion is a Congenital Simple Hamartoma of the RPE (CSRPEH). This is a benign, non-progressive, darkly-pigmented, nodular lesion with well-defined margins. Associated complications can include retinal traction, exudation, dilated feeder vessels and pigmented vitreous cells although none of these appear in this case. Fundus autofluorescence shows characteristically uniform hypofluorescence of the CSRPEH. On OCT the lesion is elevated, clearly demarcated and appears highly reflective with complete posterior shadowing. As these lesions are typically non-progressive, the prognosis is generally good. The following flow-chart from Ly A, Nivison-Smith L, Hennessy M, Kalloniatis M. (2015) Pigmented lesions of the retinal pigment epithelium. Optom Vis Sci. 92(8):844-57 is a useful reference in the differential diagnosis of pigmented RPE lesions. 
CFEH Facebook Case #1
Case #1 PDF Download
For the original question click here
Answer
Sickle-Cell Retinopathy The area scanned in the right eye and the area marked 2 in the left show peripheral neovascularization with fibrosis. There is a sunburst lesion in the left eye (lesion marked 1). The vitreous opacities seen inferiorly on the right Optomap images are consistent with old vitreous haemorrhages. This patient was subsequently referred to a retinal specialist for management. Sickle cell retinopathy results from a mutant gene that codes for the amino acids contained in haemoglobin. This causes the red blood cells to deform into a “sickle” shape when oxygen pressure is low. Sickle cell retinopathy is most prevalent in the African American population. Sunburst lesions are localized areas of retinal pigment hypertrophy, hyperplasia and pigment migration as seen in the OCT. The lesions often appear speculated and are typically found in the peripheral retina in a perivascular location. The “sunburst” lesions usually form secondary to reabsorbed sub-retinal haemorrhages and appear on fundus autofluorescence as an area of hypo-autofluorescence with a hyper-autofluorescent border. They are characteristic of sickle-cell retinopathy. Decreased visual acuity occurs due to the occlusion of parafoveal capillaries and arterioles, as well as spontaneous occlusion of the central retinal artery in some patients. This patient also has a second characteristic feature of sickle cell retinopathy – pre-retinal “sea fan” neovascularization. This typically occurs in sickle-cell patients at the posterior border of an area of retinal non-perfusion, and can lead to vitreous haemorrhage and eventually tractional retinal detachment. Other possible clinical findings typical of sickle cell retinopathy include the segmentation of blood in the blood vessels of the conjunctiva or the small surface vessels of the optic disc.

